作者: 徐永勳 醫師
校稿: Ian YC Chen, MD
上次校閱: 2018/03/27















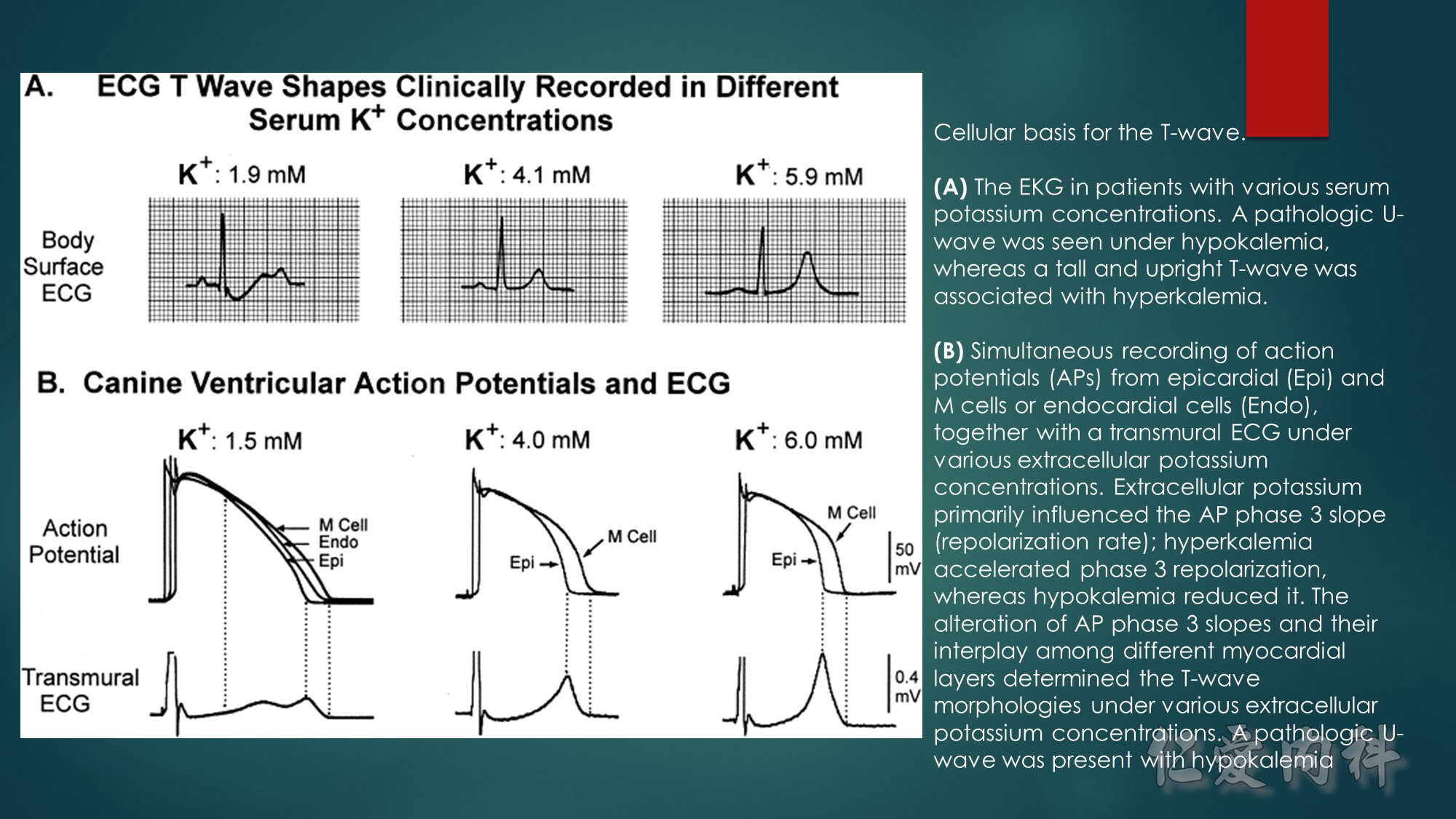
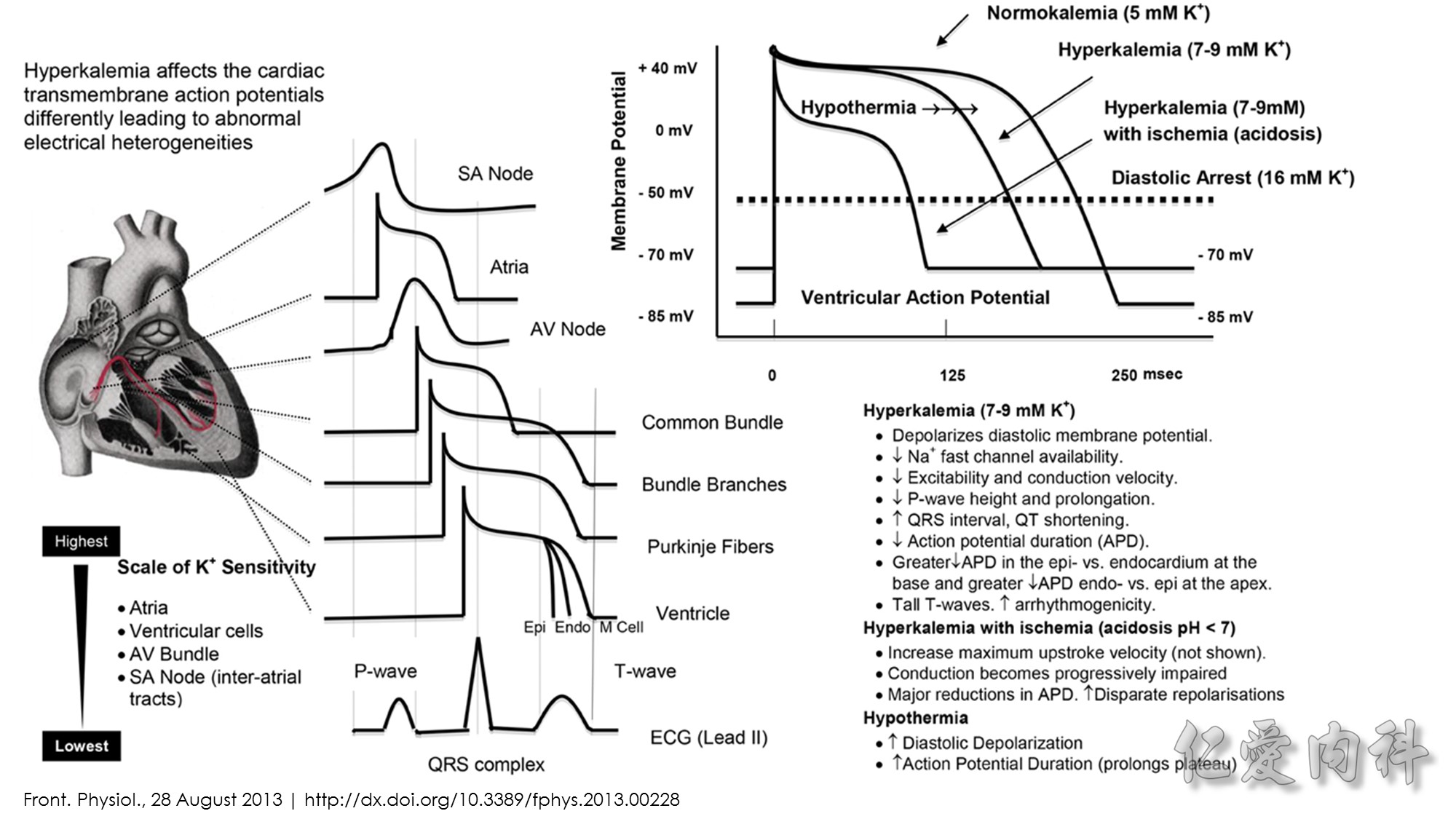
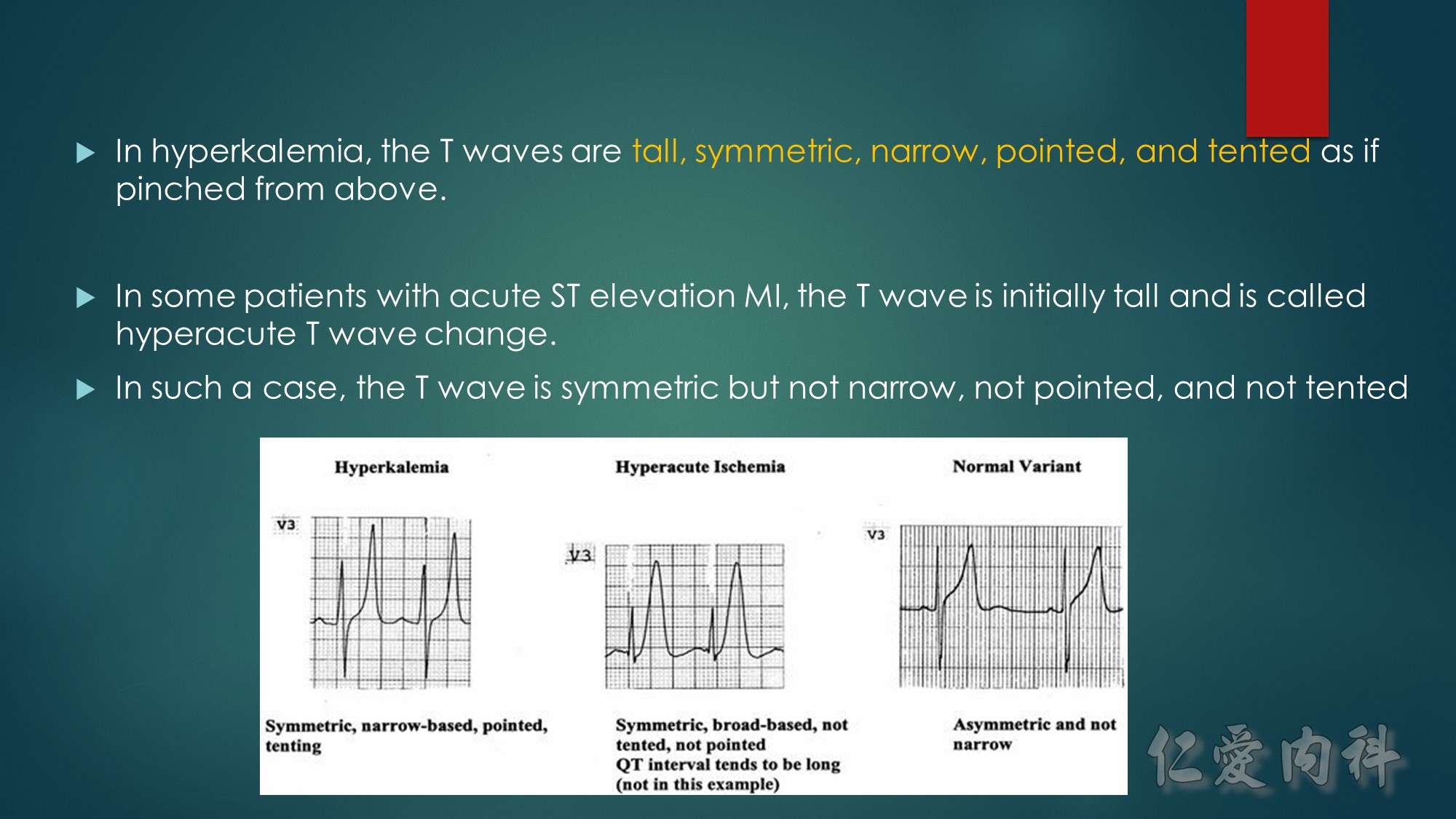
J Am Coll Cardiol. 2003;42(3):401-409







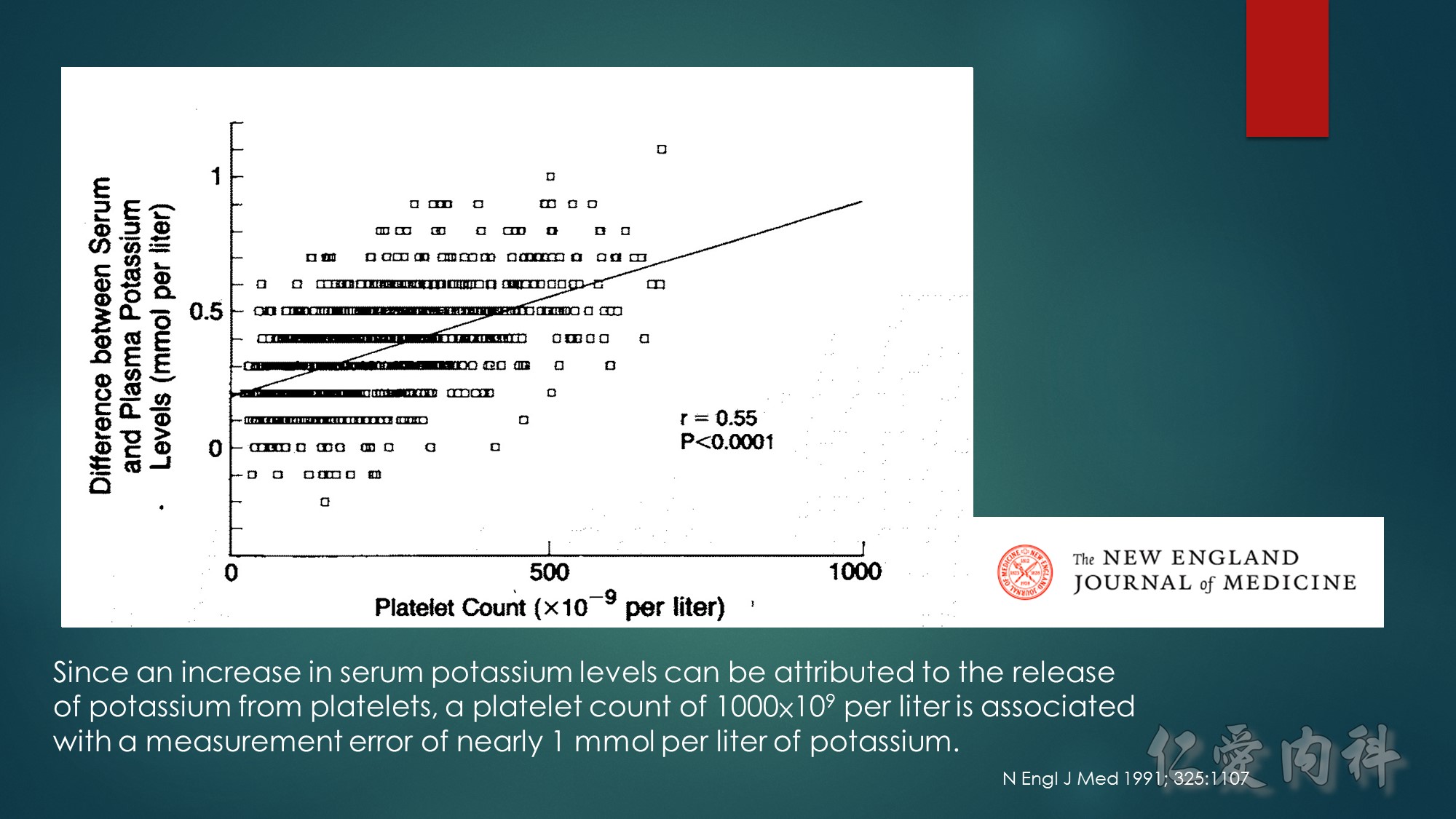
The mechanism of this phenomenon is yet to be clearly characterized, but several observations have been made. One possibility is increased sensitivity to heparin-mediated cell membrane damage during processing and centrifugation in the context of hematologic malignancy. In one study, the degree of increase in potassium was directly related to the amount of heparin contained within the tube into which the sample was collected. Mechanical stressors have also been implicated. Pneumatic tube transportation systems may lead to falsely elevated plasma potassium levels. These findings are not surprising given that the cells in patients with CLL are both fragile, and thus more susceptible to lysis, and more numerous, which can lead to significant abnormal laboratory results that may not otherwise be appreciated.


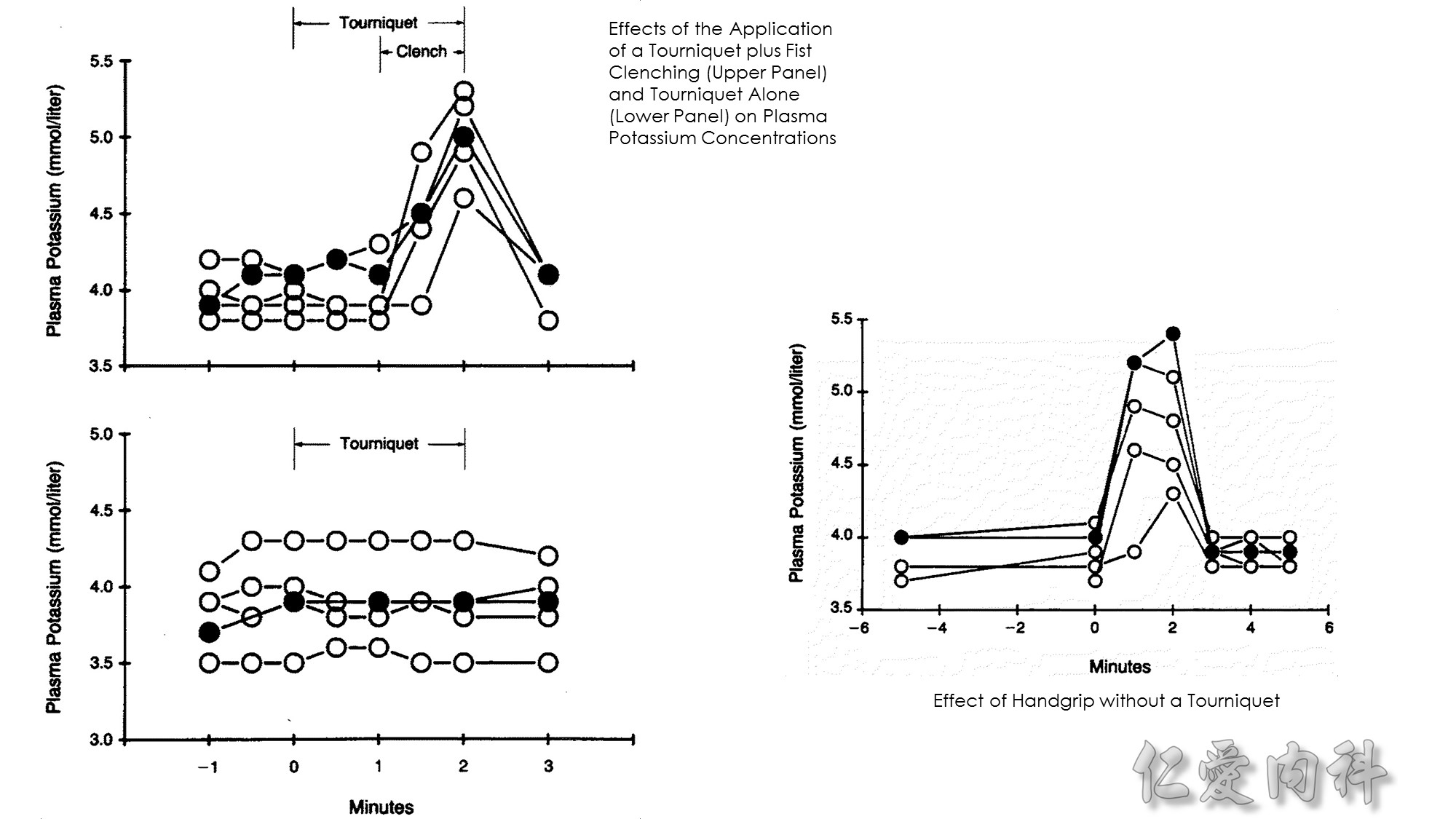
Effect of Handgrip without a Tourniquet
Two blood samples were obtained from each arm five minutes apart before the handgrip maneuver was performed. Thereafter, using a dynamometer, the subjects maintained a constant handgrip for two minutes at 30 percent of their predetermined maximal gripping strength, then relaxed the hand. Blood samples were obtained from both the test and contralateral arms at one-minute intervals during the handgrip maneuver and for three minutes thereafter




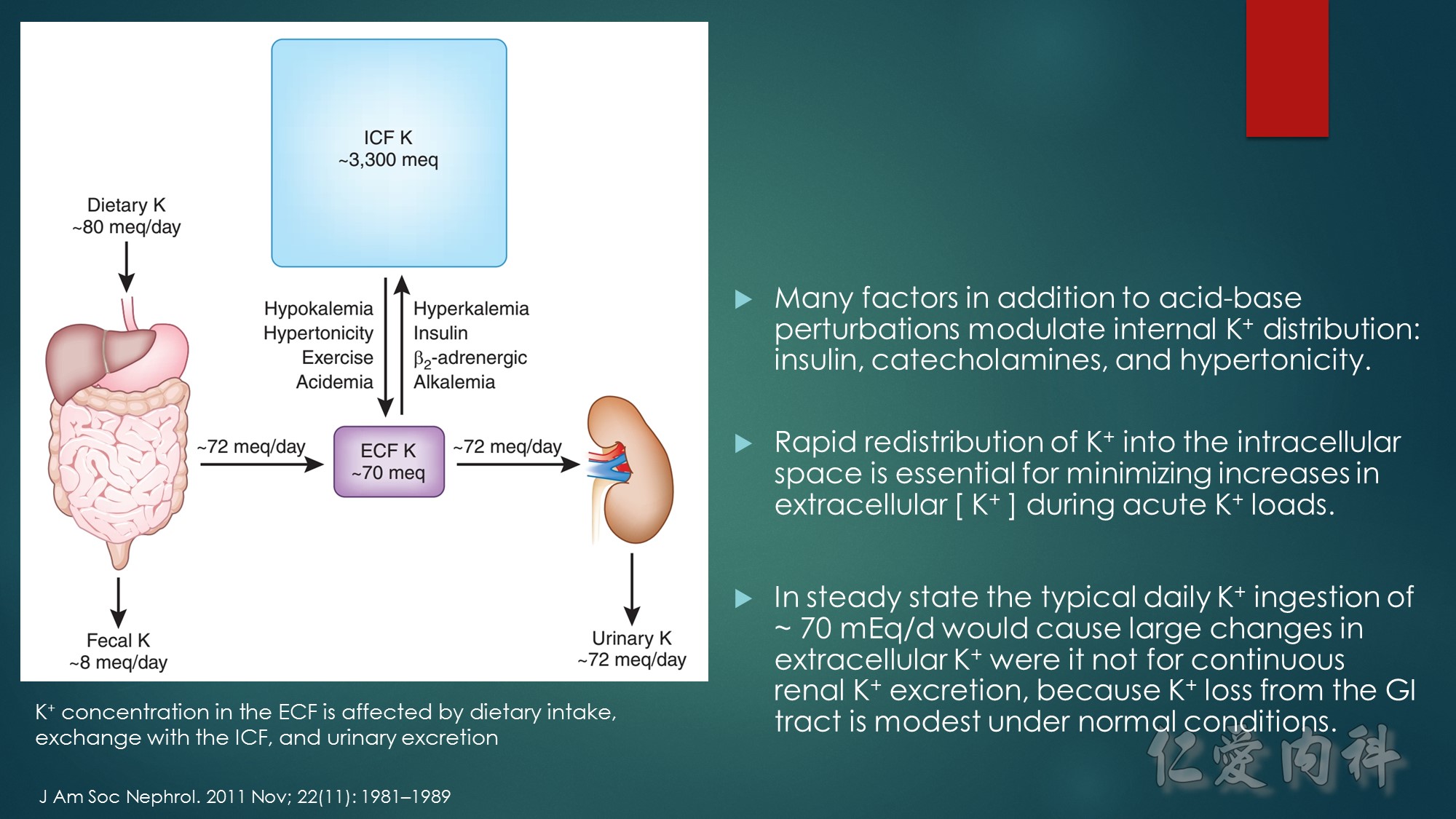
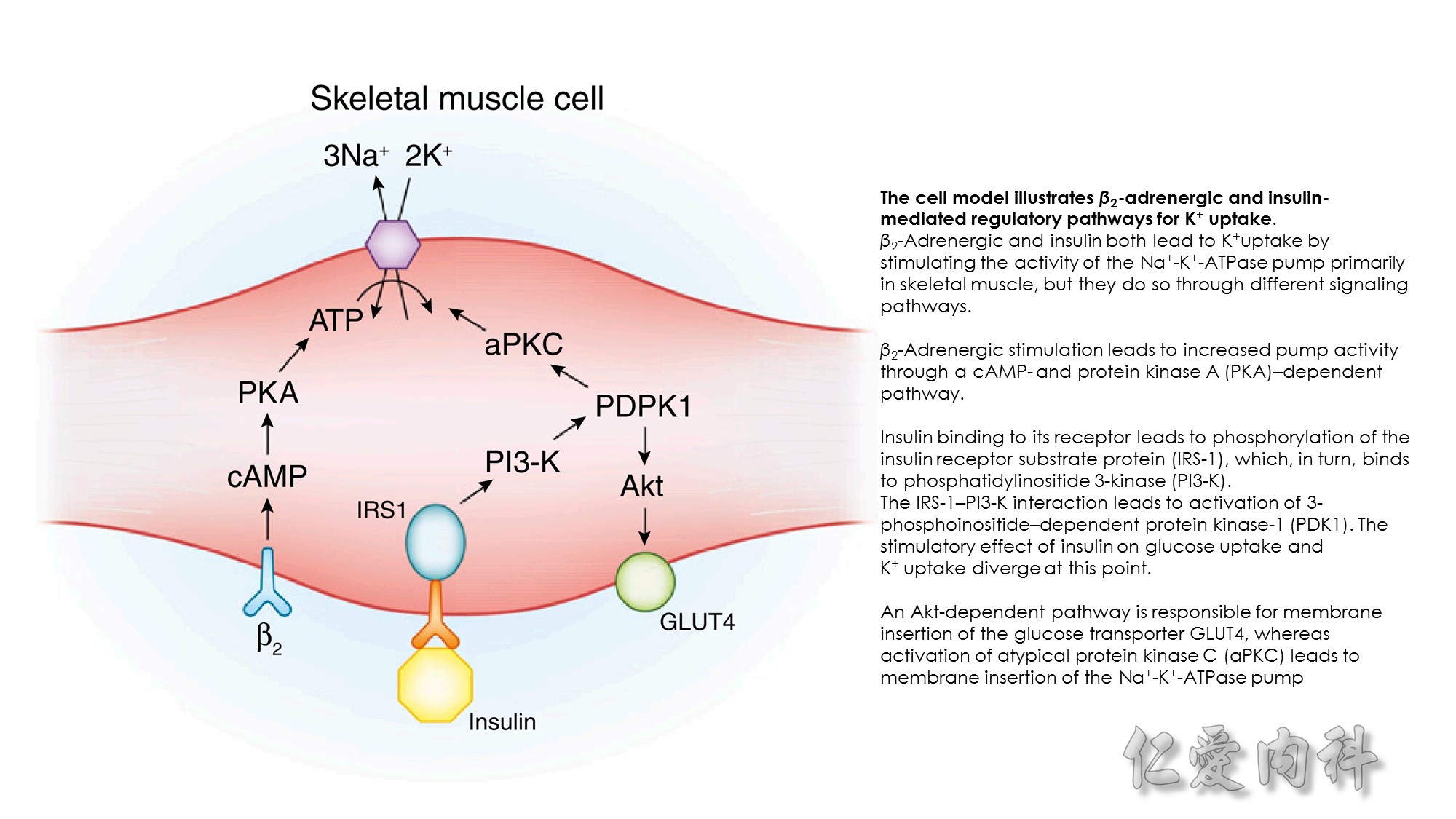
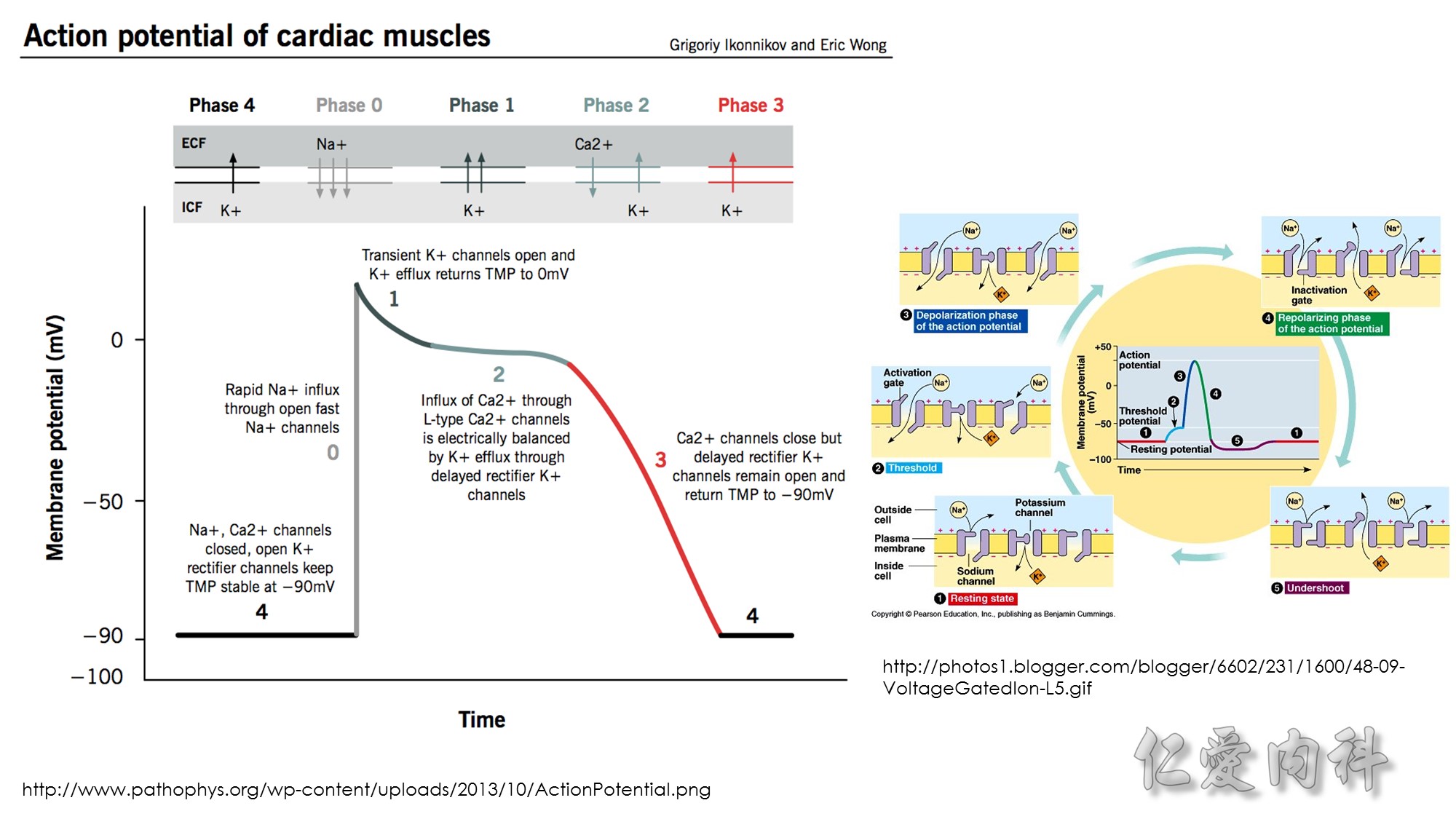
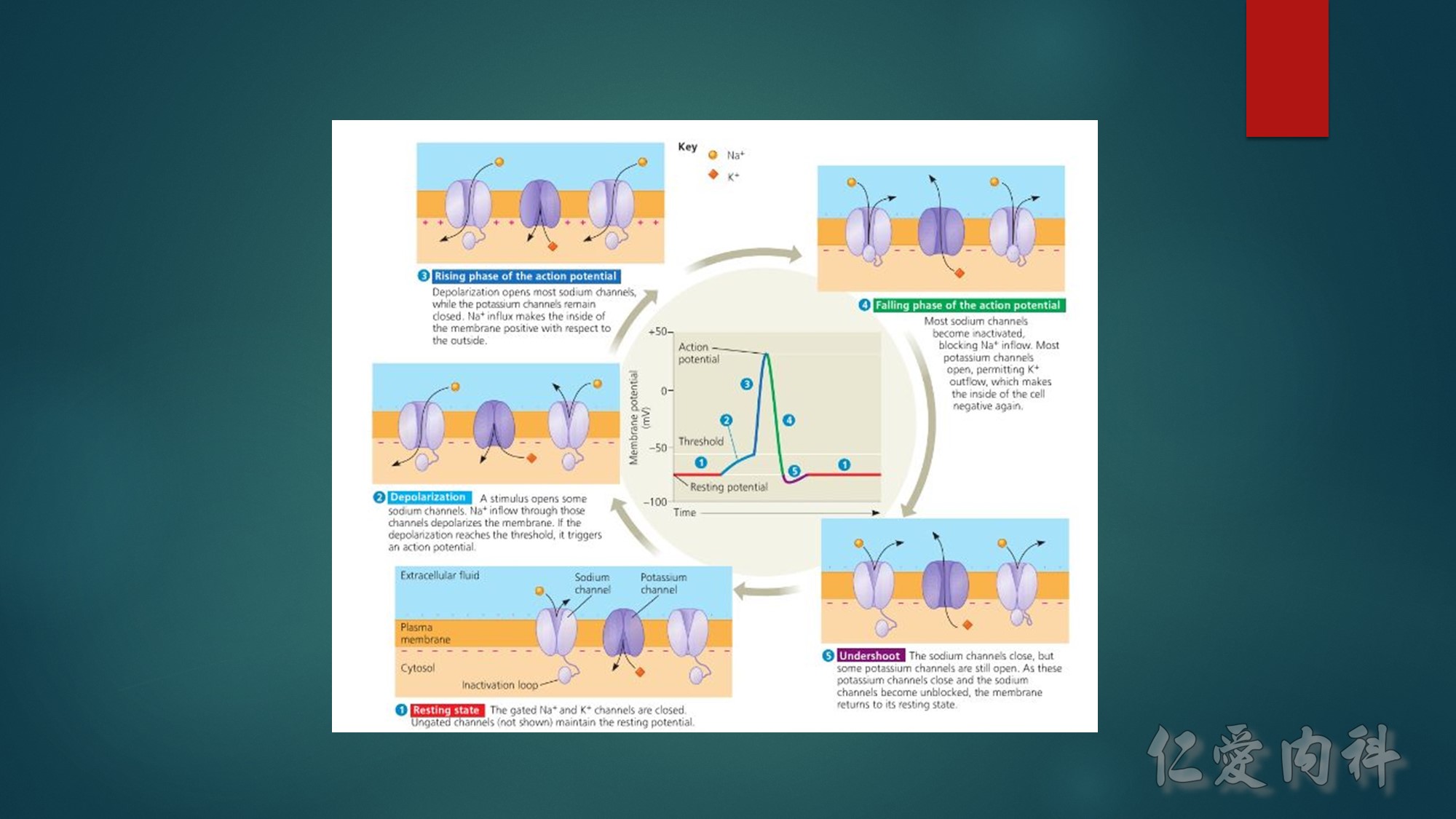
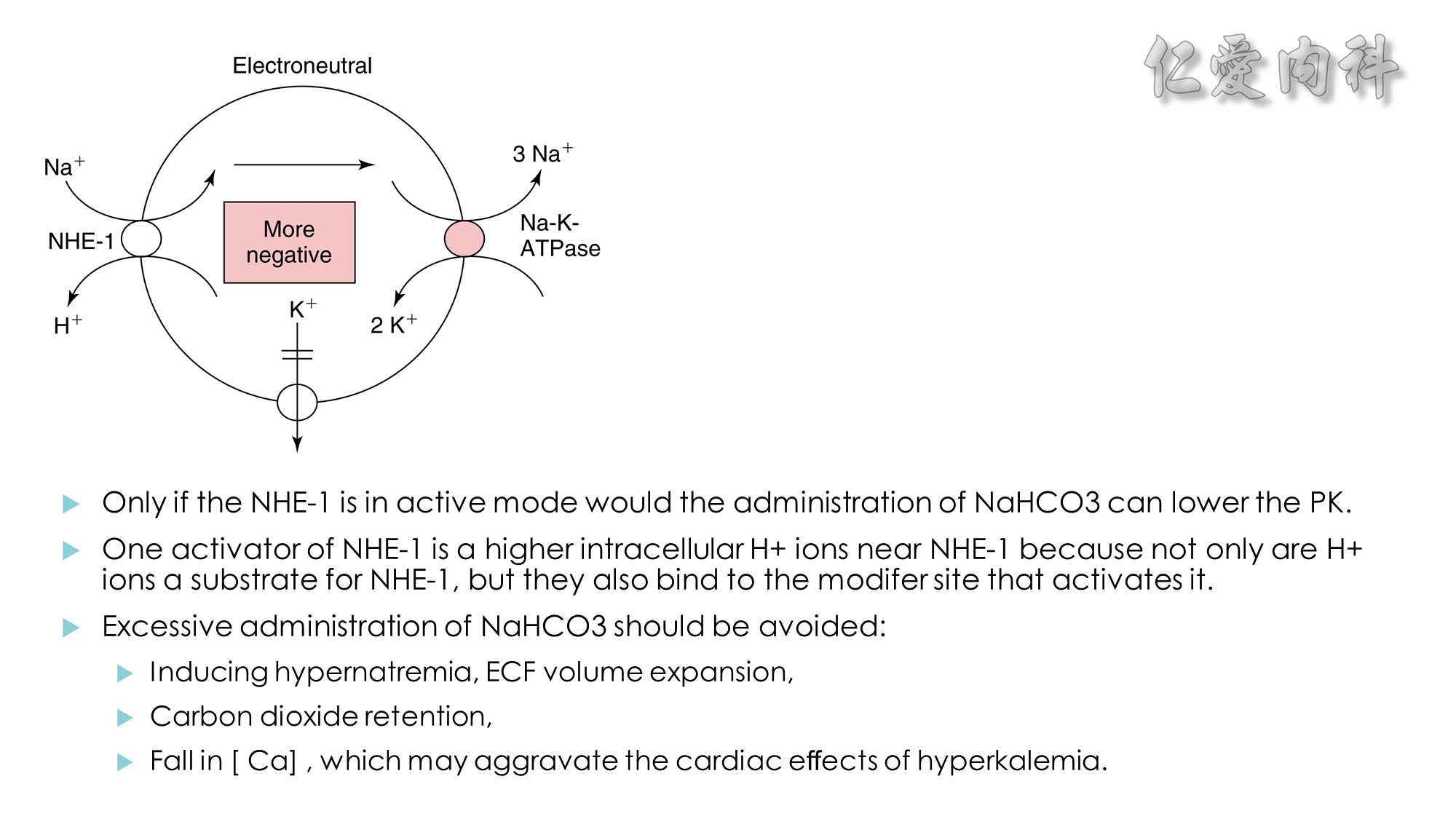
Figure 13-3 Effect of Electroneutral Versus Electrogenic Entry of Na+ Ions on the Negative Voltage in Cells.
As shown on the left side of the figure, if the source of Na+ ions pumped out of cells is Na+ ions that entered cells via the Na+ ion channel, the voltage in cells becomes less negative (count the charges), and K+ ions exit the cells if a sufficient number of open K+ ion channels are present in the cell membrane. In contrast, a more negative voltage in cells is generated by NaK-ATPase, if the source of Na+ ions pumped out is either Na+ ions that exist in cells or Na+ ions that entered the cells in an electroneutral fashion via the Na+/H+ exchanger (NHE-1); K+ ions are retained in cells (right side of the figure).
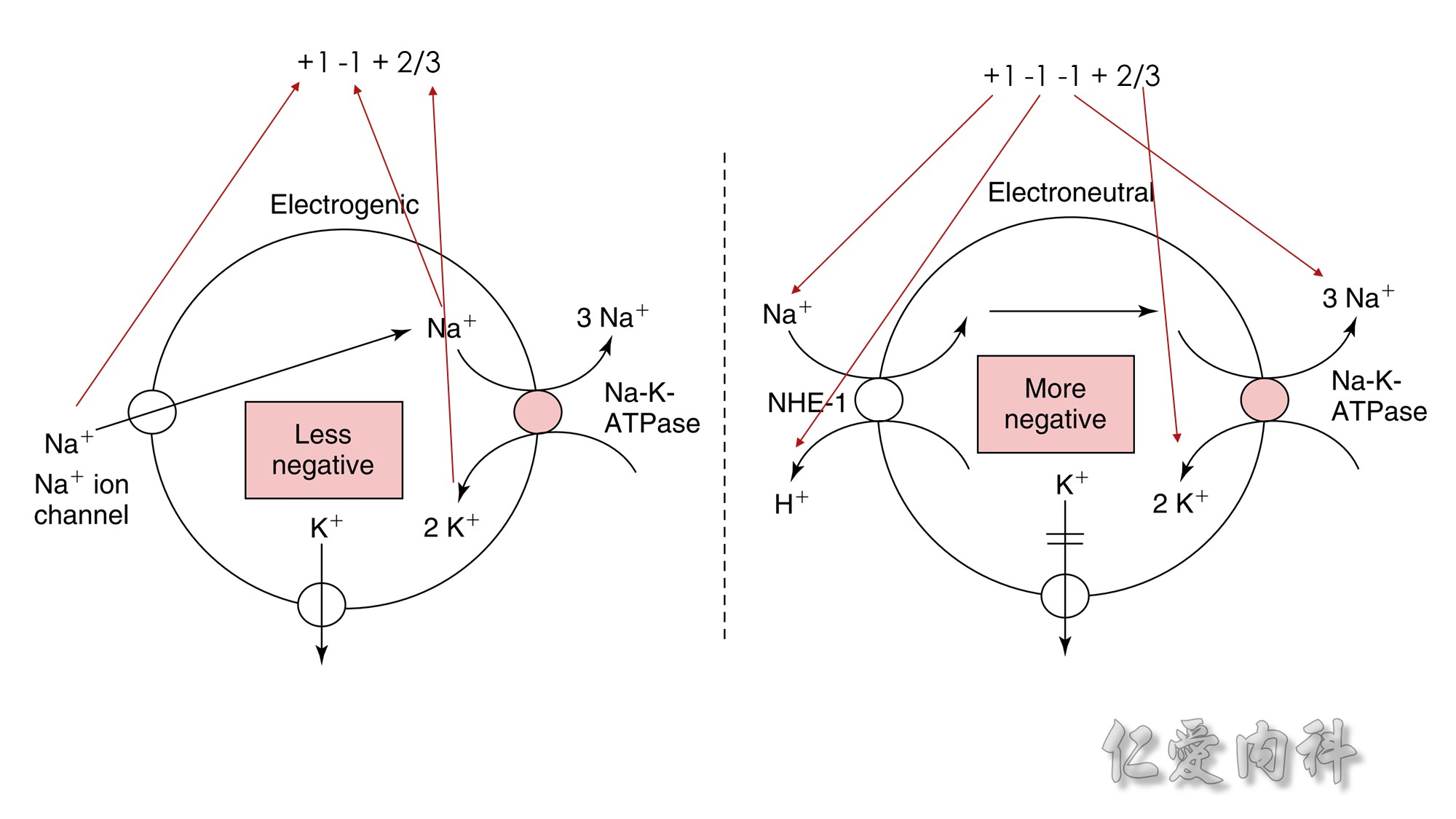 Figure 13-3 Effect of Electroneutral Versus Electrogenic Entry of Na+ Ions on the Negative Voltage in Cells.
Figure 13-3 Effect of Electroneutral Versus Electrogenic Entry of Na+ Ions on the Negative Voltage in Cells.
As shown on the left side of the figure, if the source of Na+ ions pumped out of cells is Na+ ions that entered cells via the Na+ ion channel, the voltage in cells becomes less negative (count the charges), and K+ ions exit the cells if a sufficient number of open K+ ion channels are present in the cell membrane. In contrast, a more negative voltage in cells is generated by NaK-ATPase, if the source of Na+ ions pumped out is either Na+ ions that exist in cells or Na+ ions that entered the cells in an electroneutral fashion via the Na+/H+ exchanger (NHE-1); K+ ions are retained in cells (right side of the figure).
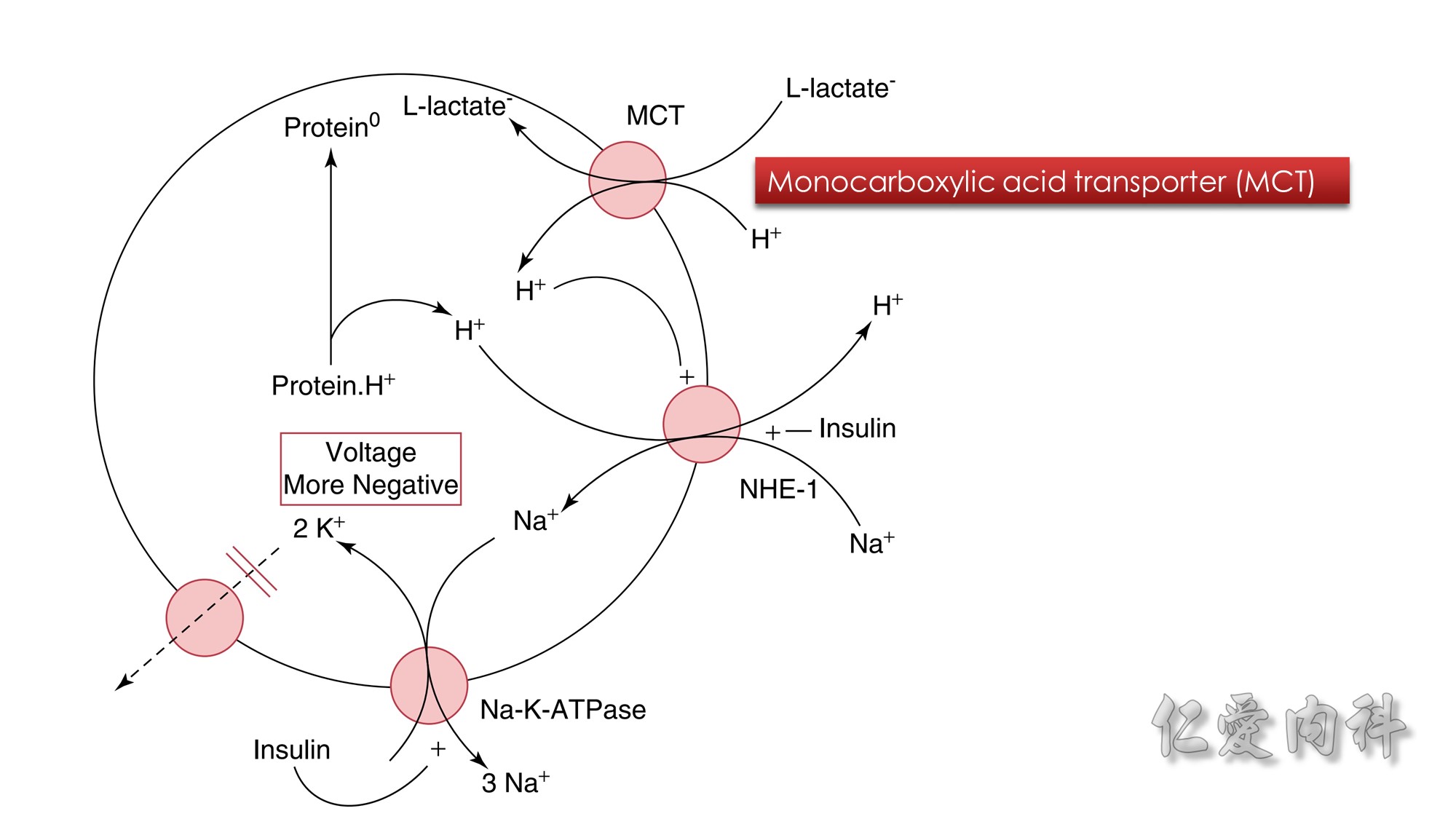
Figure 13-4 Combined Actions of Insulin and L-Lactic Acid on a Shift of K+ Ions into Hepatocytes.
The circle represents the cell membrane of hepatocytes. When L-lactic acid enters hepatocytes on the monocarboxylic acid transporter (MCT), this may lead to the release of H+ ions immediately adjacent to the NHE-1. NHE-1 is activated when H+ ions bind to its modifier site.
As a result, Na+ ions enter cells in exchange for H+ ions. The source of H+ ions is H+ ions that are bound to intracellular proteins (Protein.H+). The resulting higher concentration of Na+ ions in these cells drives the exit of Na+ ions through the Na-K-ATPase.
This leads to a higher negative intracellular voltage and hence the retention of K+ ions in hepatocytes. This process requires the presence of insulin, which, in addition to stimulating NHE-1, increases the activity and number of Na-K-ATPase in the cell membrane.
Protein°, Intracellular proteins with fewer bound H+ ions
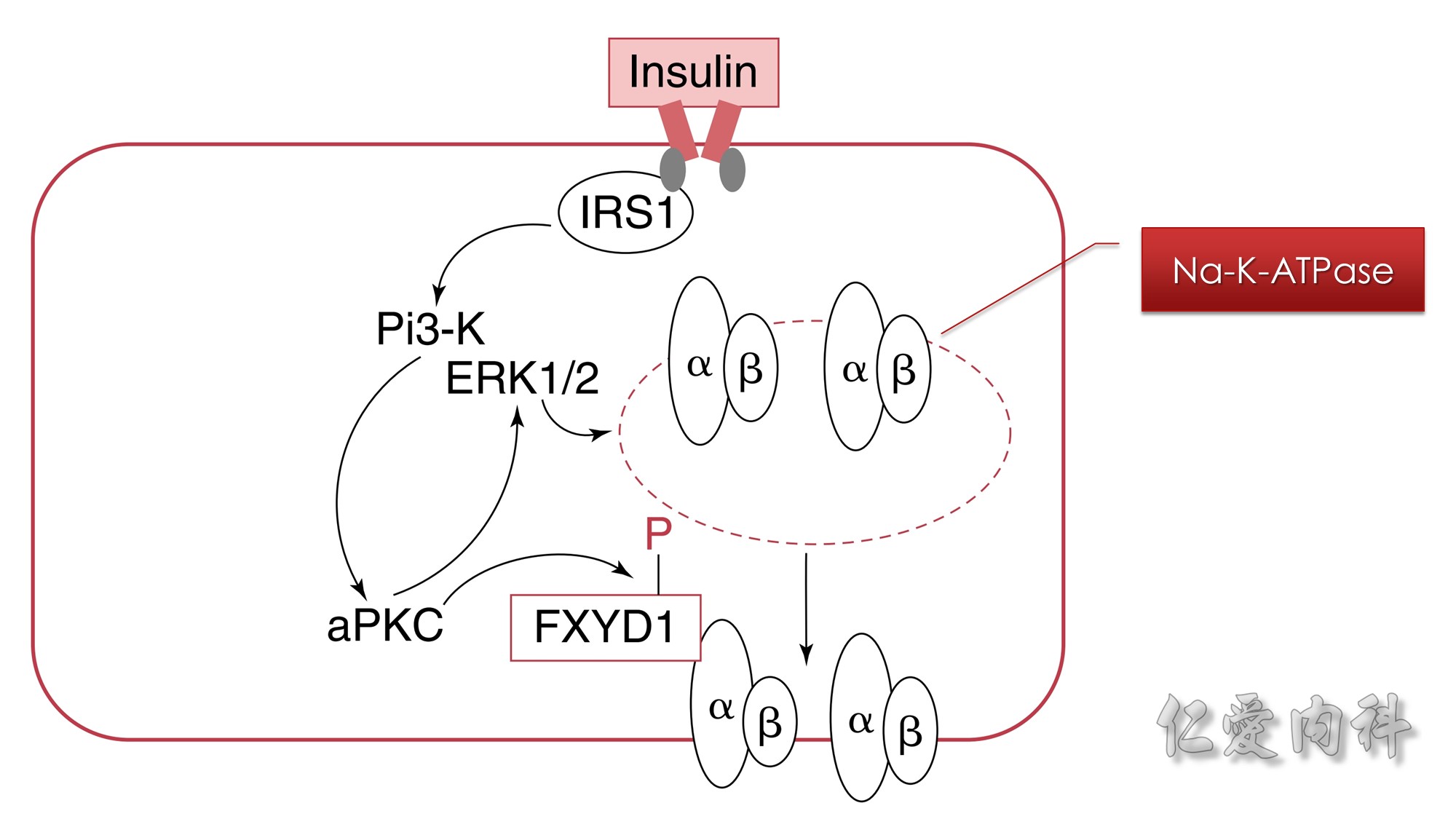 Figure 13-5 Mechanism of the Effect of Insulin to Cause a Shift of K+ Ions into Cells.
Figure 13-5 Mechanism of the Effect of Insulin to Cause a Shift of K+ Ions into Cells.
The rectangle represents a cell membrane. In addition to its effect to activate NHE-1 and the electroneutral entry of Na+ ions into cells, insulin causes phosphorylation of FXYD1 (indicated by the letter P in the figure) via atypical protein kinase C (aPKC). Insulin also increases the expression of Na-K-ATPase in cell membranes. This effect is mediated through phosphoinositide 3-kinase (Pi3-K) and extracellular signal regulated kinases 1 and 2 (ERK1/2).
ERK1/2 kinases induce phosphorylation of the α subunit of the Na-K-ATPase, which promotes the translocation of Na-K-ATPase from an intracellular pool to the cell membrane.
The vertically oriented, side-by-side ovals labeled α and β represent the Na-K-ATPase.
Insulin binds to its cell surface receptor. This causes activation of insulin receptor substrate (ISR-1). This in turn activates phosphoinositide 3-kinase(Pi3-K). Activated Pi3-K phosphorylates thereby activates atypical protein kinase C (aPKC). This has two effects. First, activated aPKC phosphorylates FXYD1. Second, it also activates the extracellular signal regulated kinases 1 and 2 (ERK1/2) to phosphorylate the alpha-subunit of Na-K-ATPase in the cytosol of the cell, promoting its translocation to the cell membrane. IRS1, Insulin receptor substrate 1
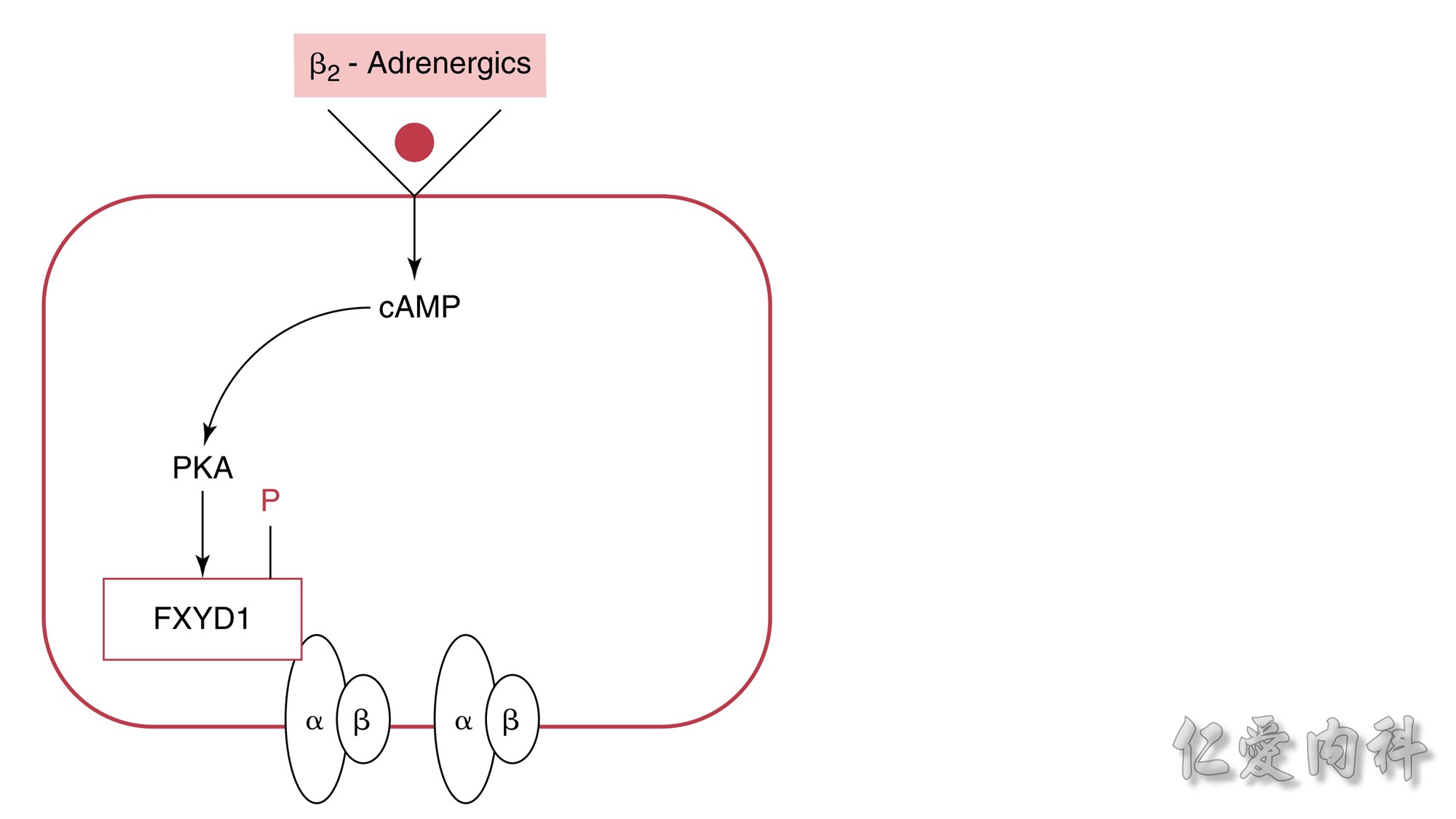
Figure 13-6 Mechanism of the Effect of β2-Adrenergic Agonists to Cause
a Shift of K+ Ions into Cells.The rectangle represents a cell membrane. β2-
Adrenergic agonists activate adenylate cyclase, and this leads to stimulation of the conversion of adenosine triphosphate (ATP) to cyclic adenosine
monophosphate (cAMP). This second messenger, in turn, activates protein kinase-A (PKA), which induces phosphorylation of the FXYD1 (indicated by
the letter P in the figure). This disrupts the interaction of FXD1 with the α-subunit of the Na-K-ATPase, which results in an increase in the affinity
of Na-K-ATPase for Na+ ions and/or its Vmax. The increase in export of preexisting intracellular Na+ ions leads to a higher negative voltage in cells and
hence a shift of K+ ions into cells.
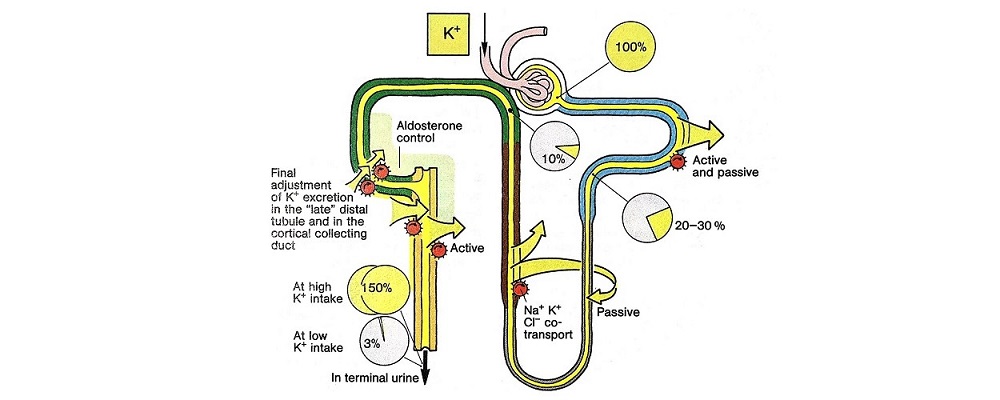
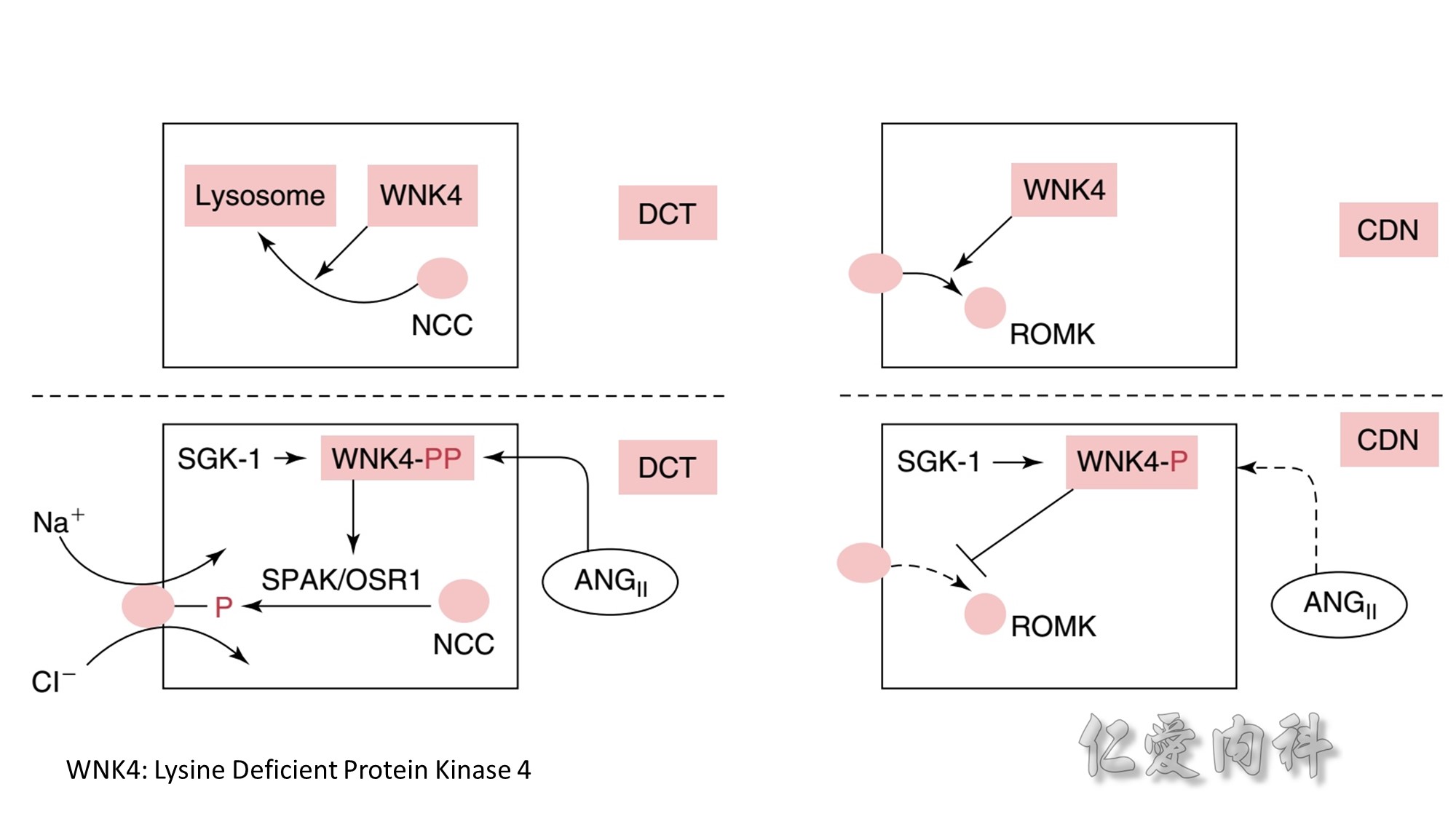
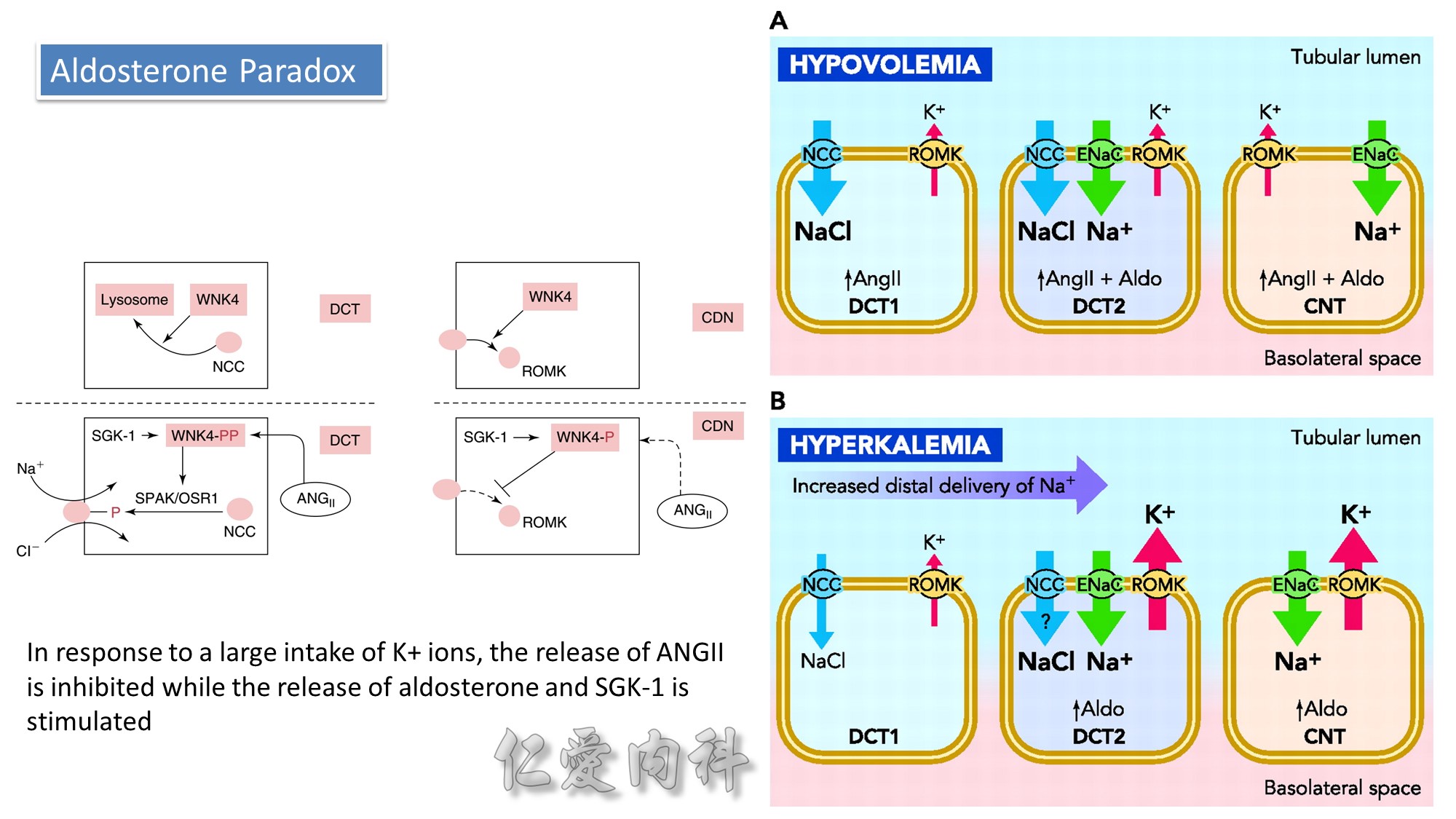
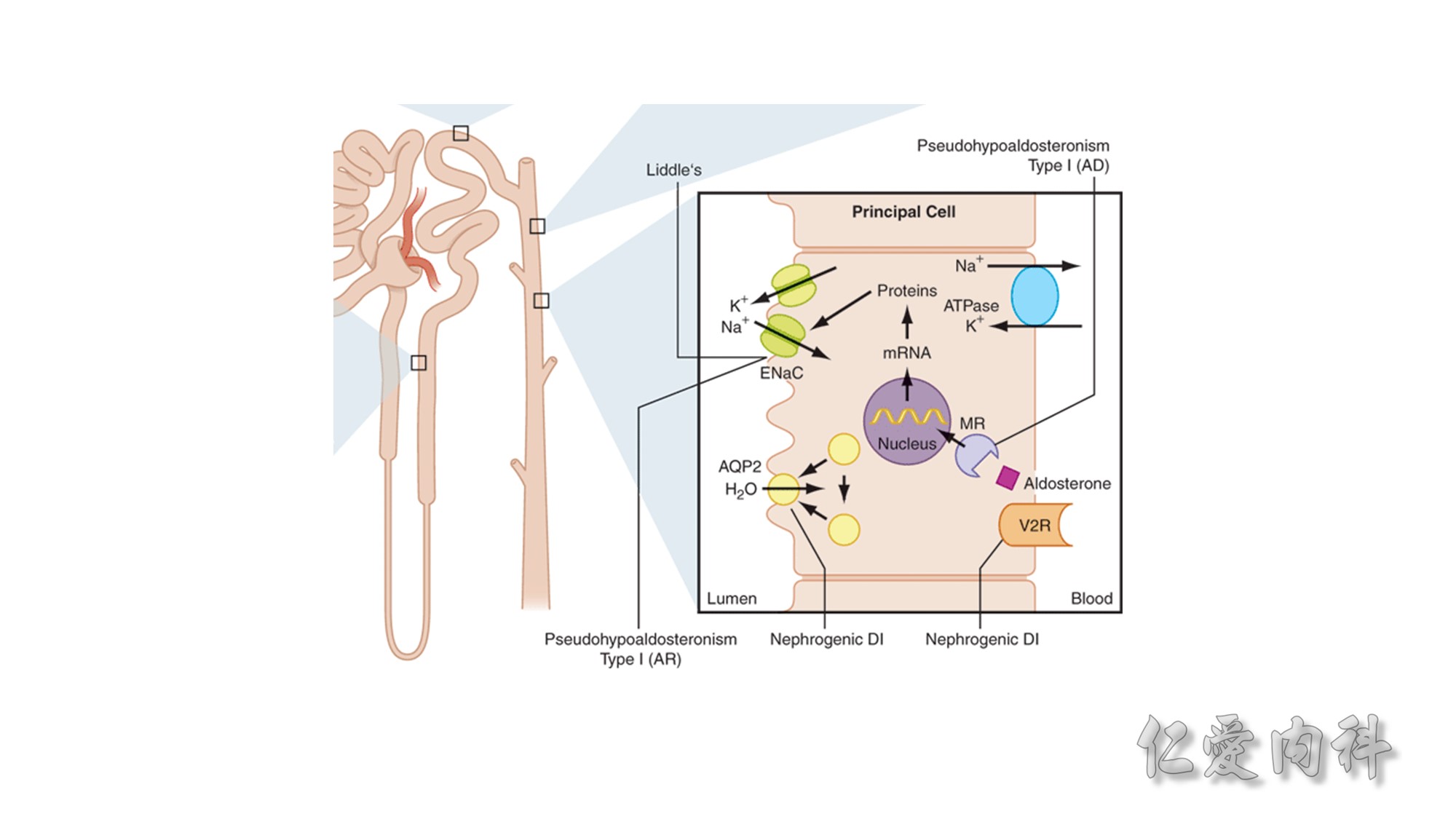
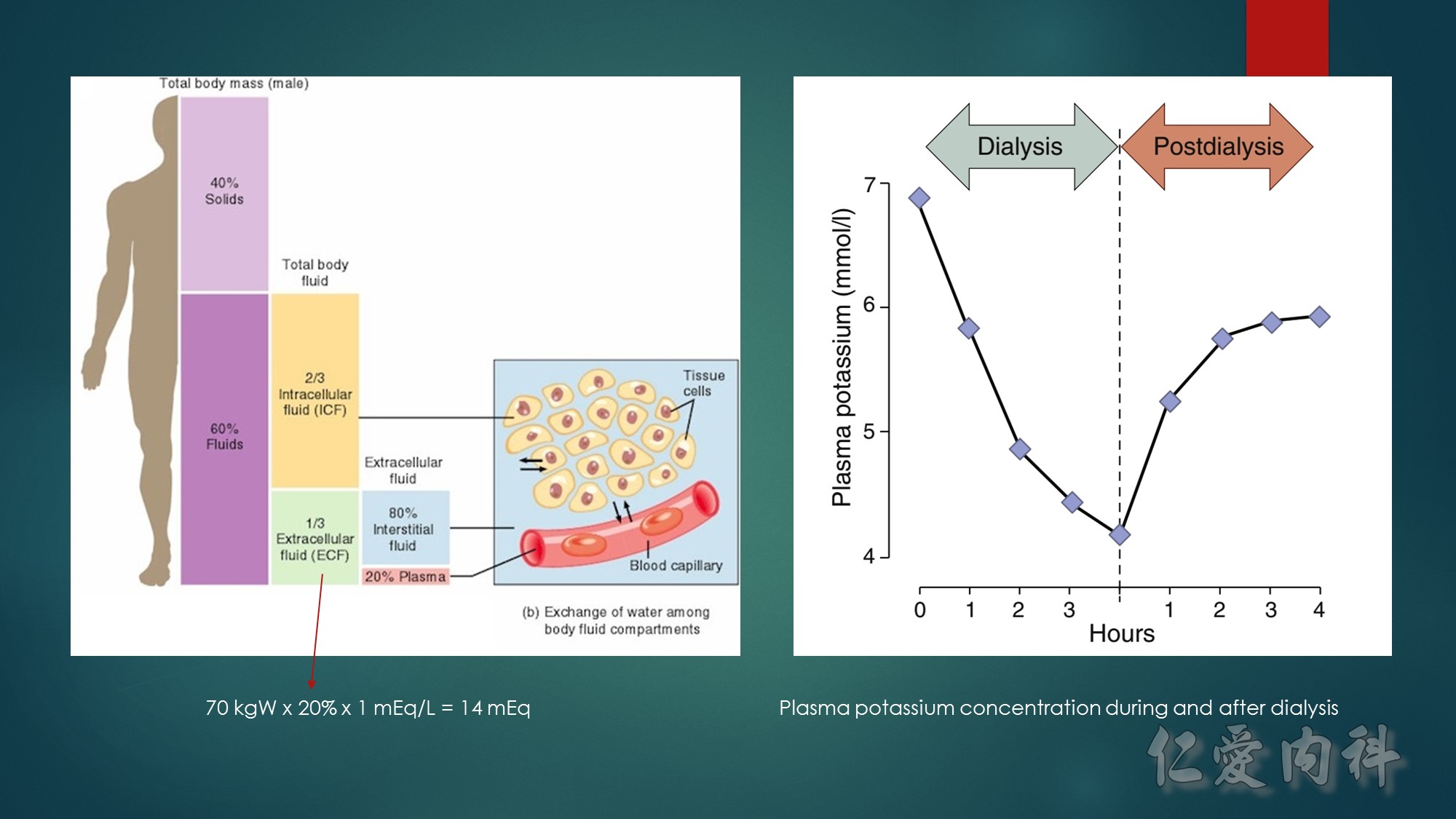
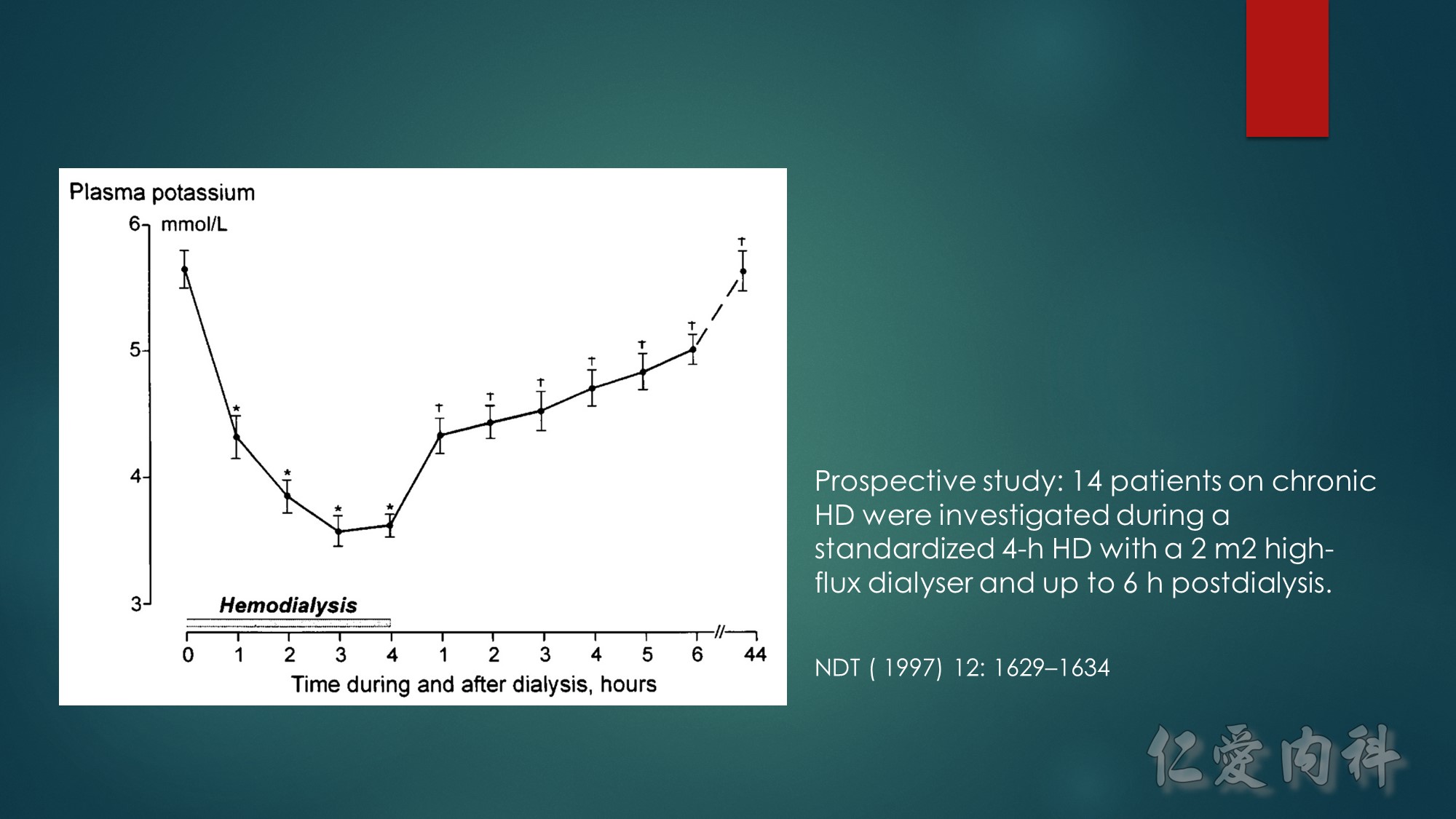
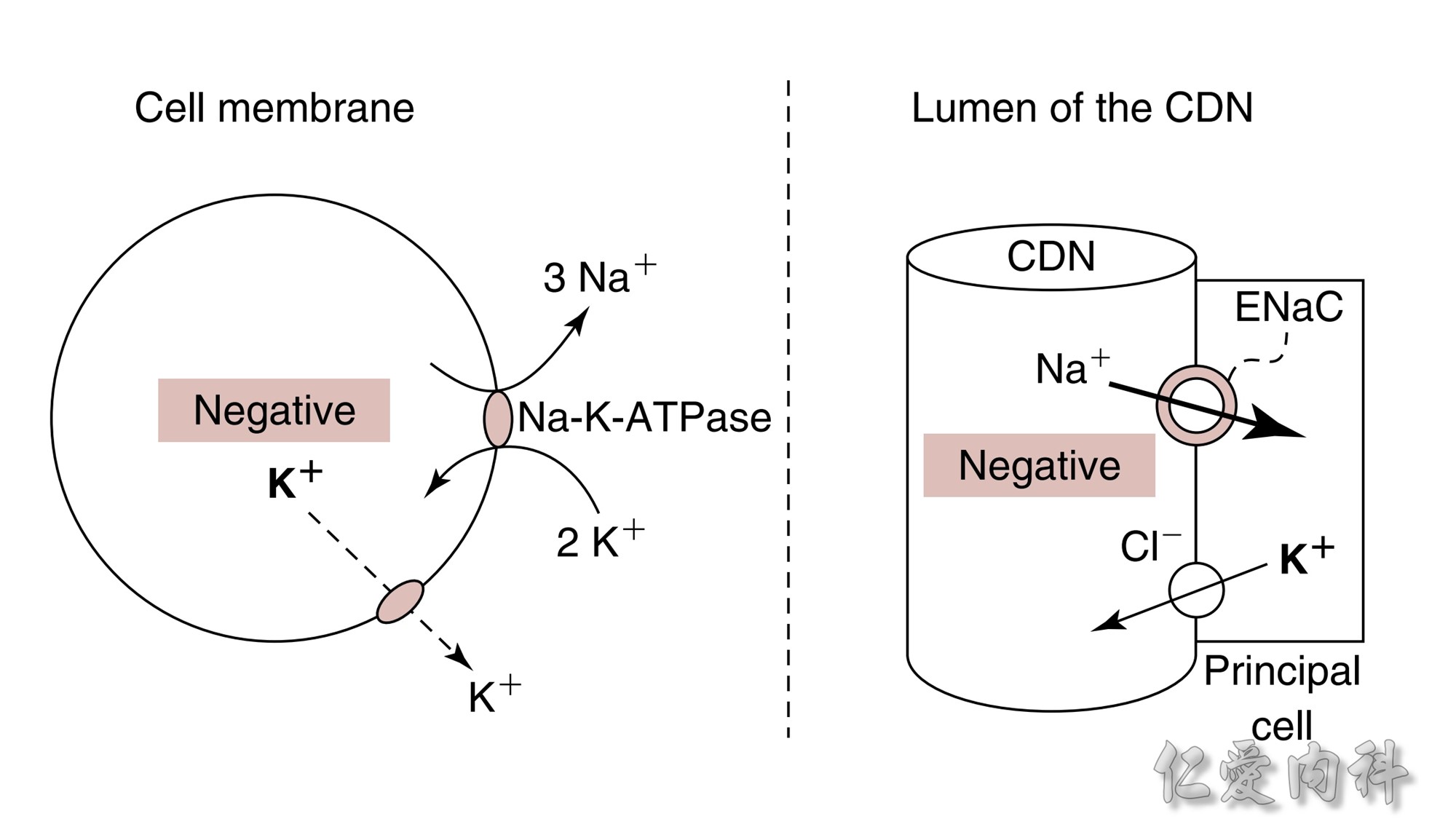 Concept for the Movement of K+ Ions Across Cell Membranes.
Concept for the Movement of K+ Ions Across Cell Membranes.
The circle on the left represents a cell membrane and the cylinder on the right represents the cortical distal nephron (CDN). There is a much higher
concentration of K+ ions in cells than in the extracellular fluid compartment because of the negative voltage inside cells, which is generated in part because more Na+ ions are extruded from than K+ ions are imported into cells by the Na-K-ATPase. To cause a redistribution of K+ ions across cell membranes, there must be a change in either the magnitude of the negative voltage inside cells or in the number, open probability, and/or conductance of K+ ion channels (because the concentration of K+ ions in cells is higher than the predicted value from the electrochemical equilibrium).
K+ ions will enter the lumen of the CDN when it has a negative voltage and if there are open renal outer medullary K+ ion channels in the luminal membrane of principal cells in the CDN.
A lumen negative voltage is generated when Na+ ions are reabsorbed in an electrogenic fashion (i.e., reabsorption of Na+ ions without their accompanying anions, which are usually Cl- ions).
Reabsorption of Na+ ions occurs via the epithelial Na+ ion channel (ENaC).

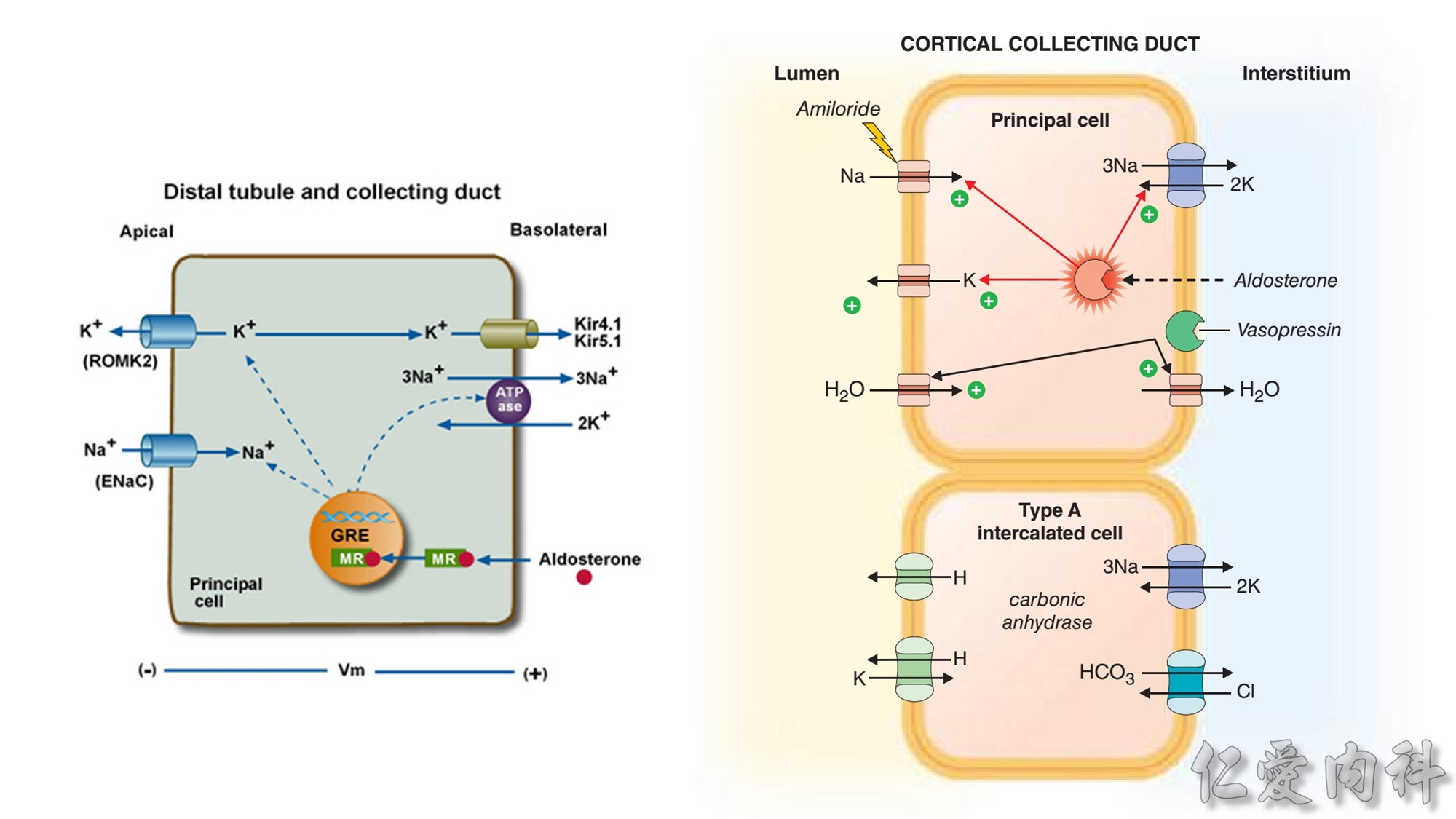
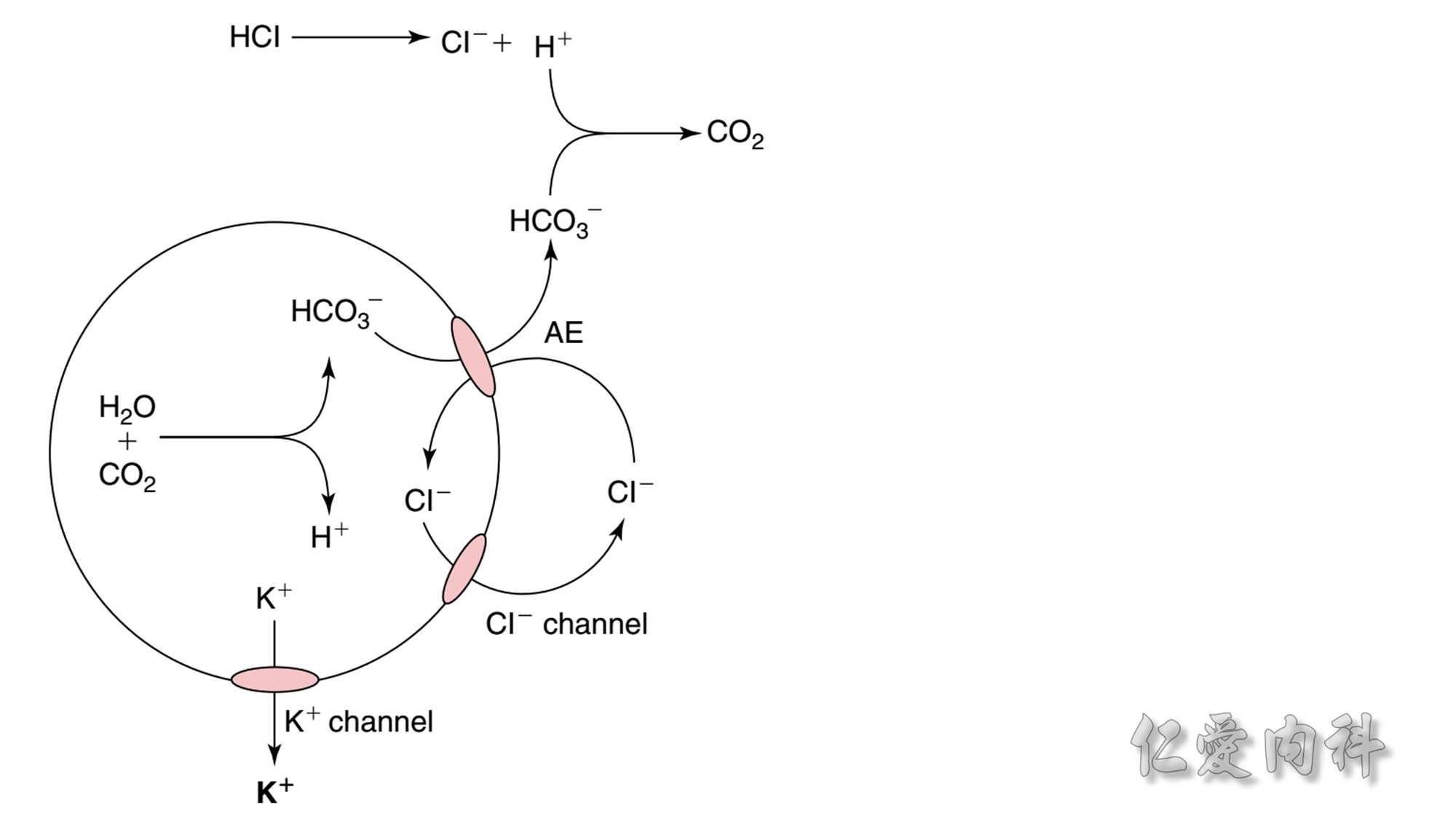 Figure 13-7 Shift of K+ Ions Out of Cells in Patients with Metabolic Acidosis Caused by the Addition of Nonmonocarboxylic Acids.
Figure 13-7 Shift of K+ Ions Out of Cells in Patients with Metabolic Acidosis Caused by the Addition of Nonmonocarboxylic Acids.
The circle represents a cell. Inorganic acids (e.g., HCl) or nonmonocarboxylic organic acids (e.g., citric acid) cannot enter cells by the monocarboxylic acid transporter.
Therefore, a mechanism is needed to permit some of these H+ ions to be titrated using HCO3- anions in the intracellular fluid compartment.
This may involve activation of the Cl-/HCO3 anion exchanger (AE) with transport of HCO3- ions out of cells and of Cl- ions into cells. Cl- ions are forced out of cells in an electrogenic fashion because of the electrochemical driving force and the presence of open Cl- ion channels in cell membranes, the voltage in these cells becomes less negative, and K+ ions exit the cells.
























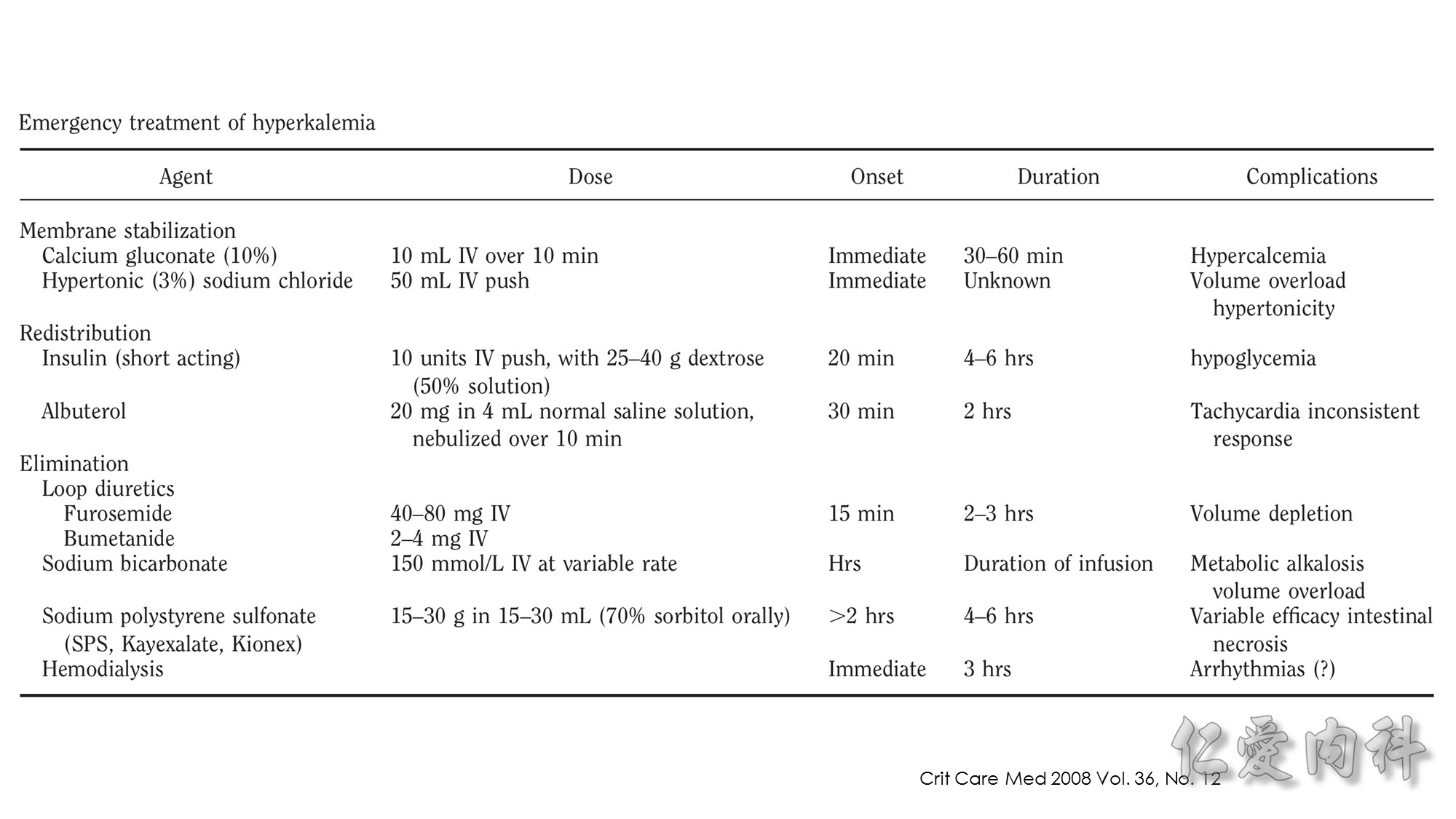
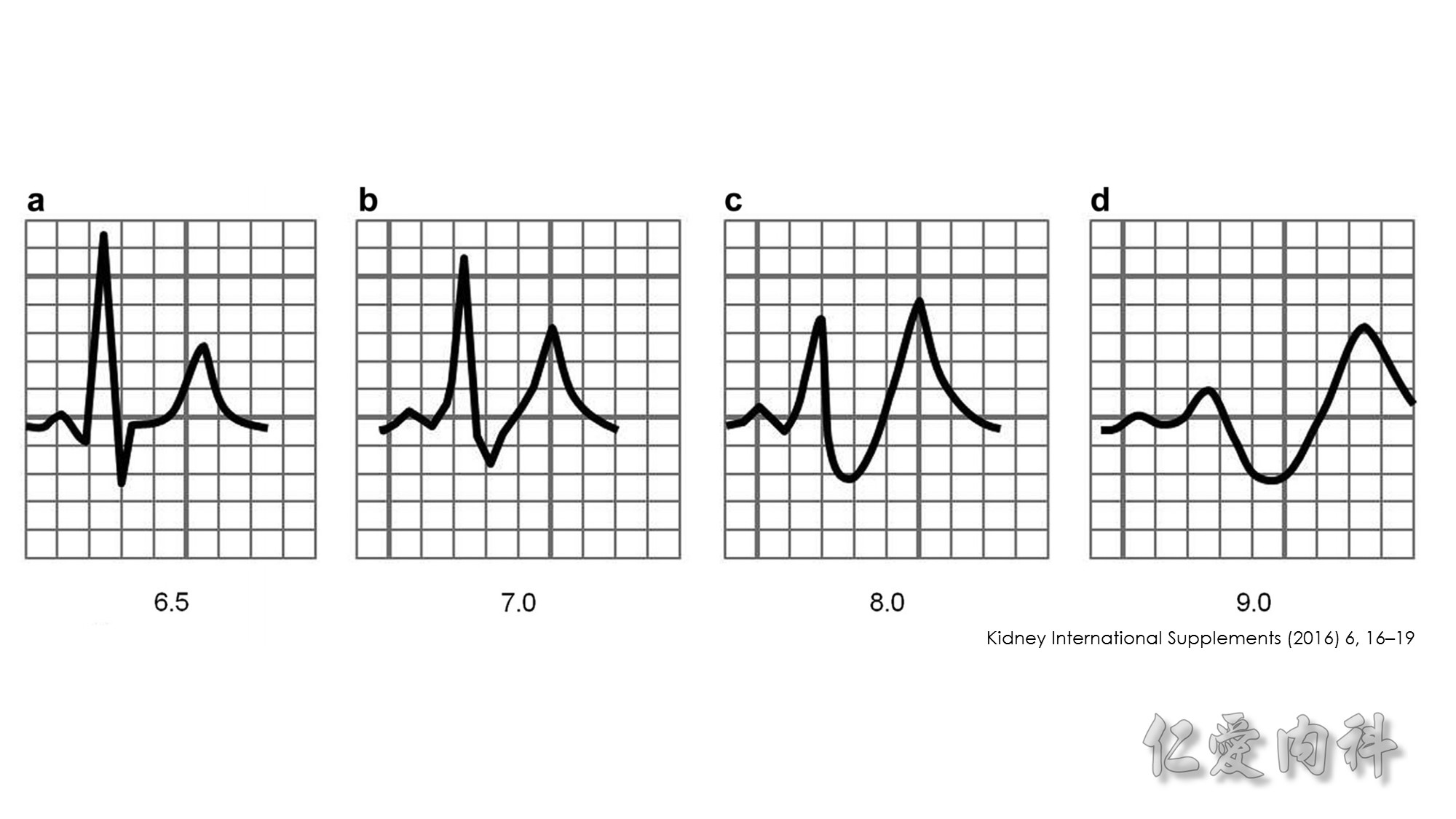
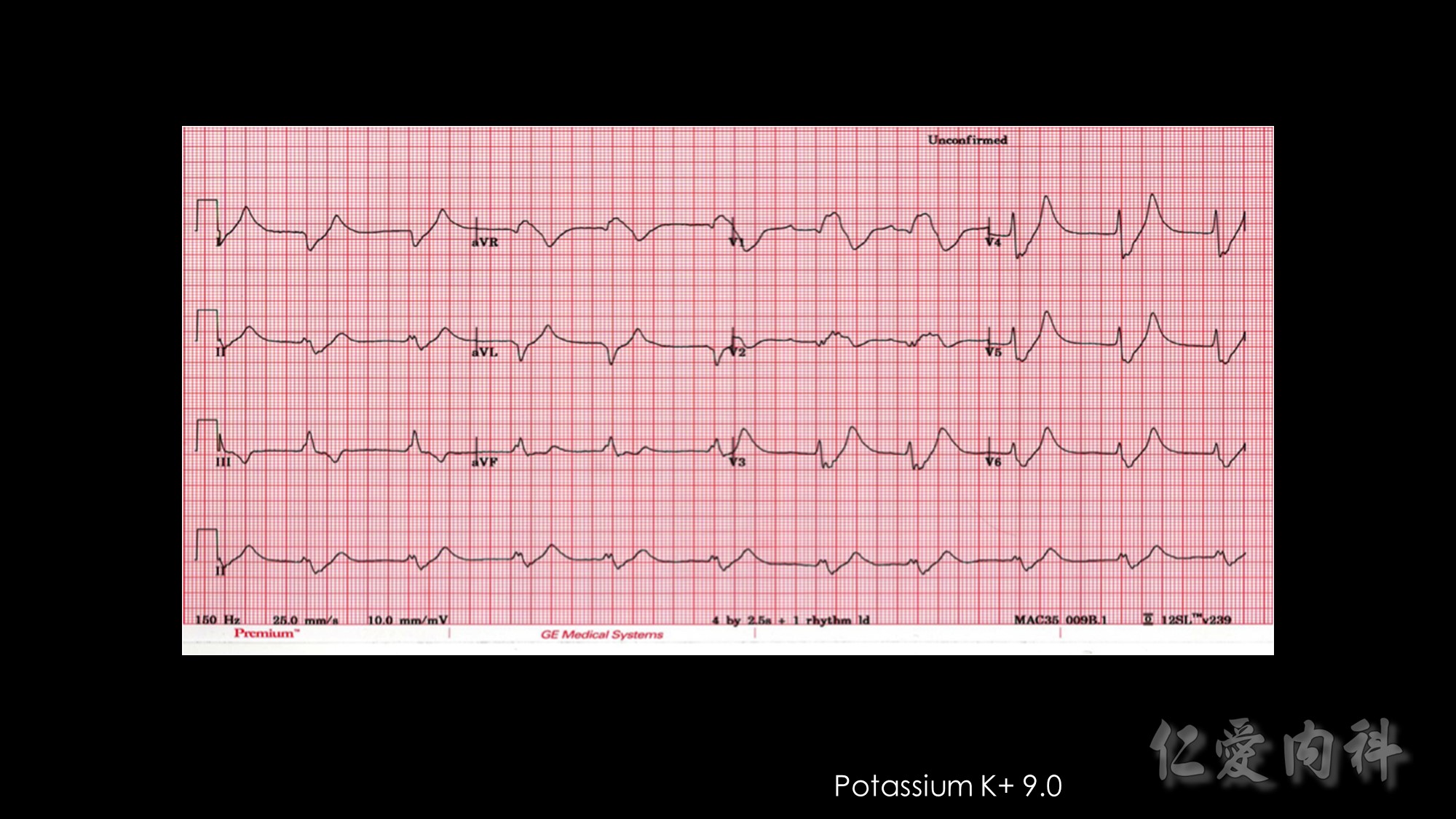
In 1964, Chamberlain reported 5 patients with serum potassium concentrations ranging from 8.6 to 10 mmol/l, illustrating “immediate” (within 5 minutes) resolution of the most advanced electrocardiographic findings after intravenous calcium. Our knowledge of when to use this intervention, or what dose and formulation (calcium gluconate or calcium chloride) to use has not advanced since these early observations.
http://medicineforresidents.blogspot.tw/2011/03/calcium-gluconate-in-digitalised-heart.html
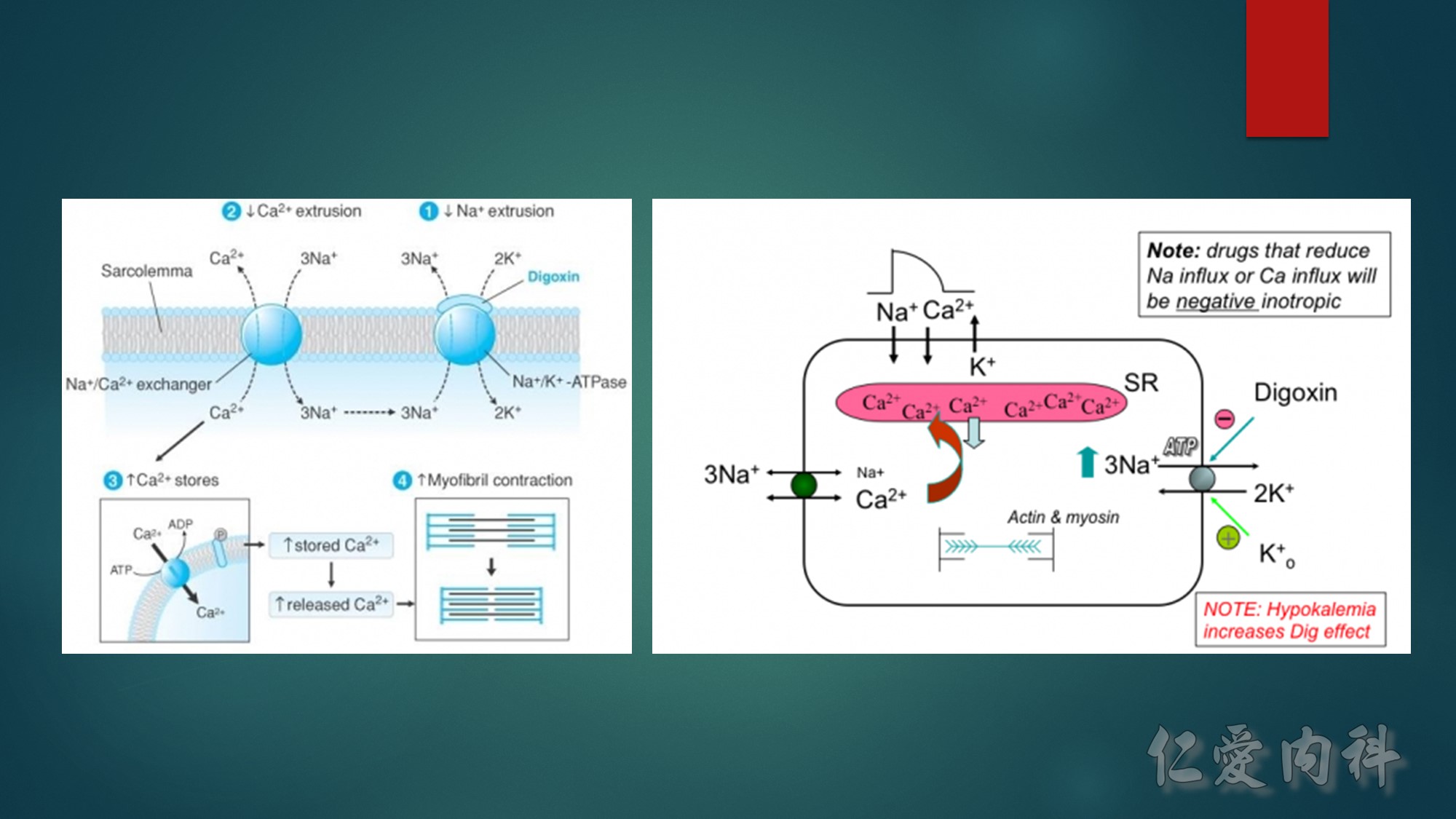
First…..the mechanism of action of Digoxin- Digoxin inhibits the Na+/K+-ATPase(exchanges 2 K for 3Na) in the cardiac myocyte by competing with potassium,and causes intracellular sodium concentration to increase. This then leads to an accumulation of intracellular calcium by blocking the Na+-Ca++ exchange system. In the heart, increased intracellular calcium causes more calcium to be released by the sarcoplasmic reticulum, thereby making more calcium available to bind to troponin-C, which increases contractility (inotropy).
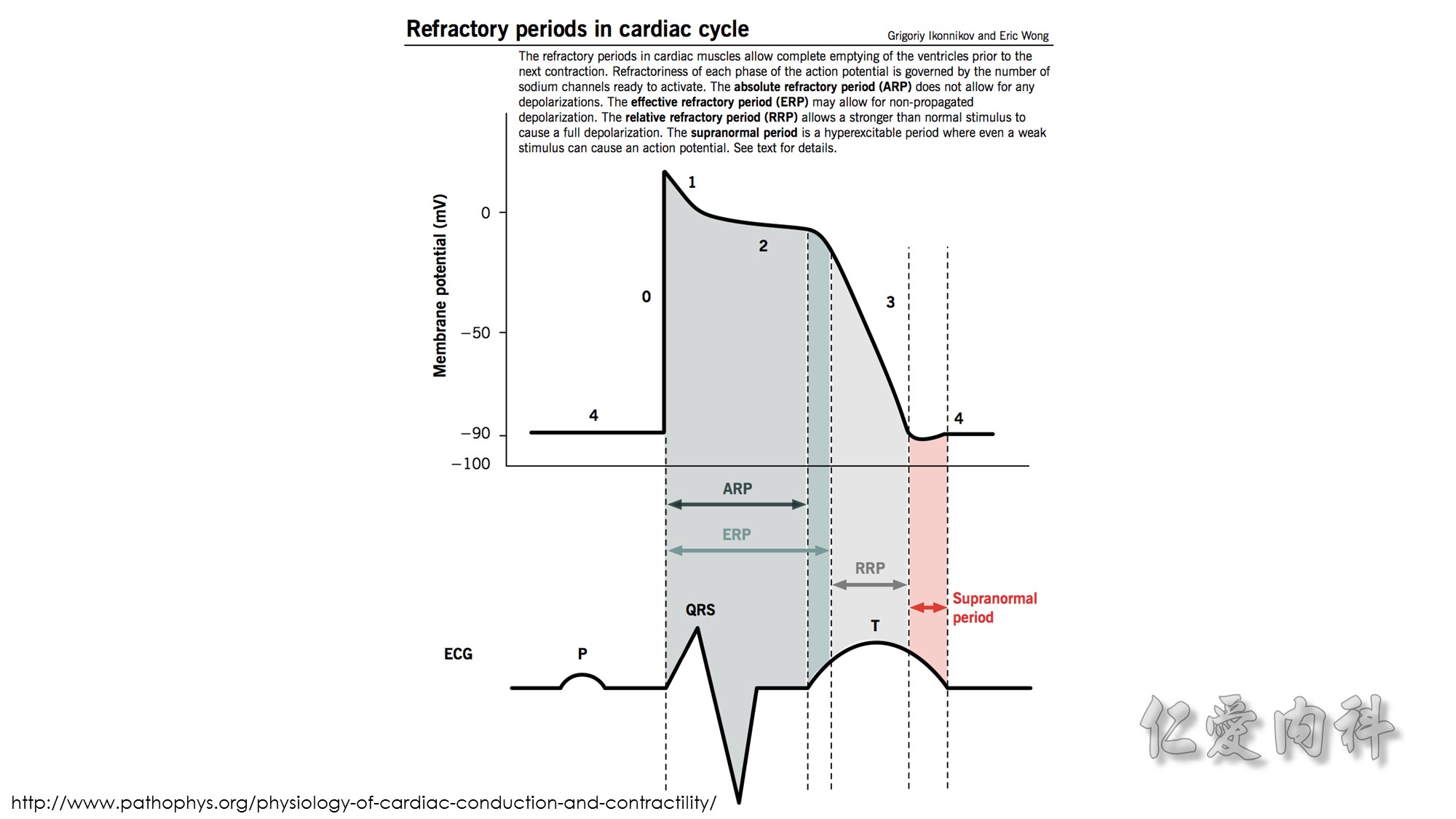
Second…..the mechanism of action of Calcium gluconate- a litlte more cellular pathology! . Normally the cardiac myocyte has a resting membrane potential(RMP is -90mV) and a threshold potential at which it is excited(TP is-75mV).So a 15mV depolarisation is needed to excite the myocyte. In hyperkalemia, the RMP becomes less negative (-80mV) , but the TP remains at -75mV. This means that ..now..only a 5mV depolarisation is enough to excite the myocyte. This is the cause of hyperexcitability leading to arrhythmia.Calcium gluconate…by increasing Ca transport across the membrane reduces the TP from -75mV to around -65mV …restoring the 15mV depolarisation needed to excite the myocyte. (the numbers in mV are just an example) …… Annals of Emer Med 2011
Sooo…in patients on Digoxin(which causes positive inotropy through calcium)…if more calcium is given….it can lead to more intracellular calcium in mycocyte leading to what has been described as cardiac tetany due to prolonged depolarisation. So ,does hyperkalemia make this process worse?? Well, probably not. ——Since K+ and digoxin compete for Na/KATPase, the binding depends on the concentration of the two. In hyperkalemic state…K+ binds preferentially than digoxin, on Na/K ATPase, and thus resulting in diminished digoxin action. (to better understand…think about hypokalemic states…where digoxin will bind preferentially to the receptor, and hence result in digoxin toxicity!–which is well known)
A slightly different scenario would be…when some one(with no K+ problems) takes too much digoxin…which will also result in digoxin binding preferentially than K+, to Na/KATP ase resulting in Dig toxicity. In this setting….pt may go hyperkalemia..as more K+ ends up extracellularly (due to not binding with Na/K ATPase), and if this patient gets Ca gluconate..then again..it can causes arrhythmias.

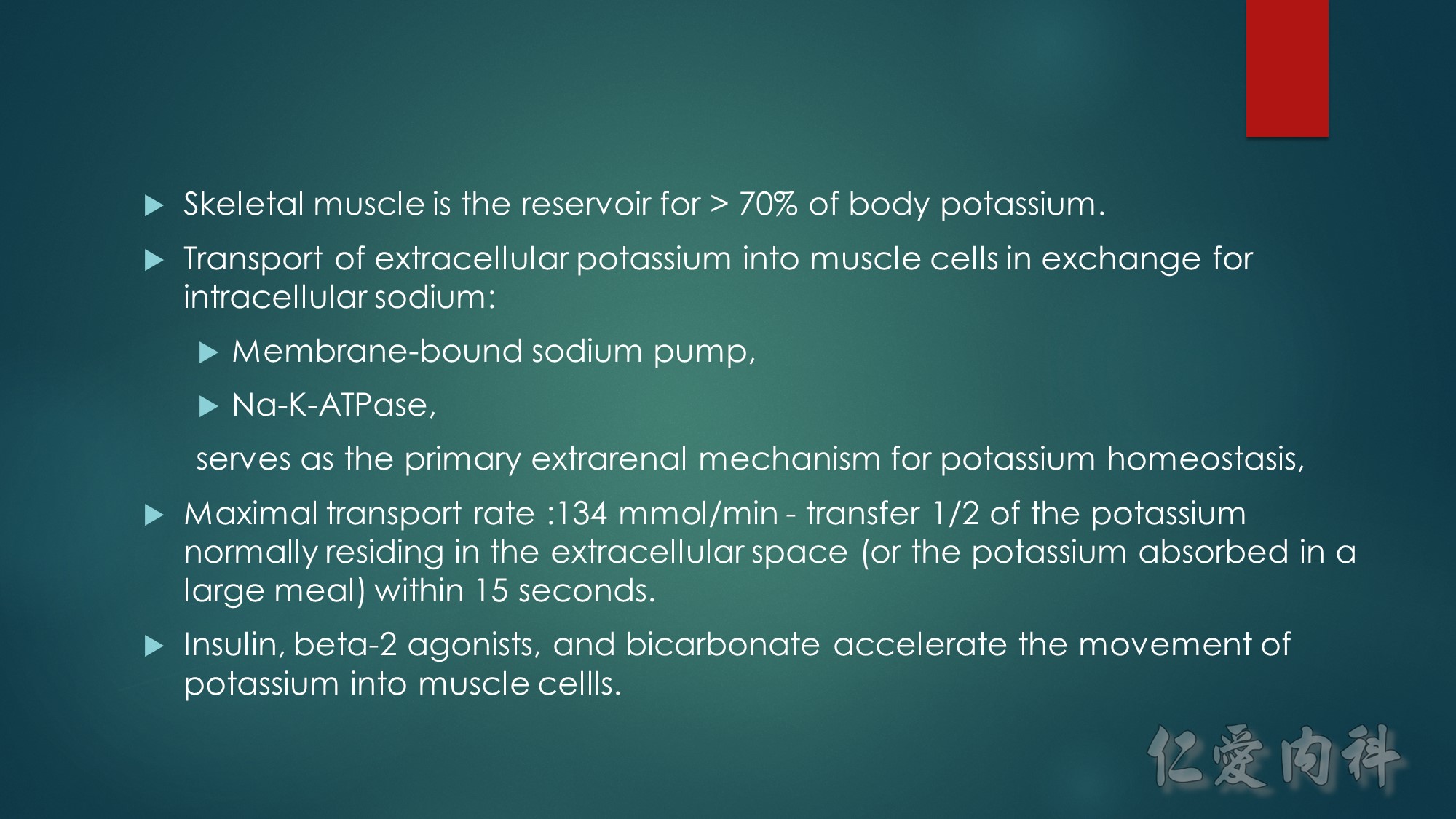








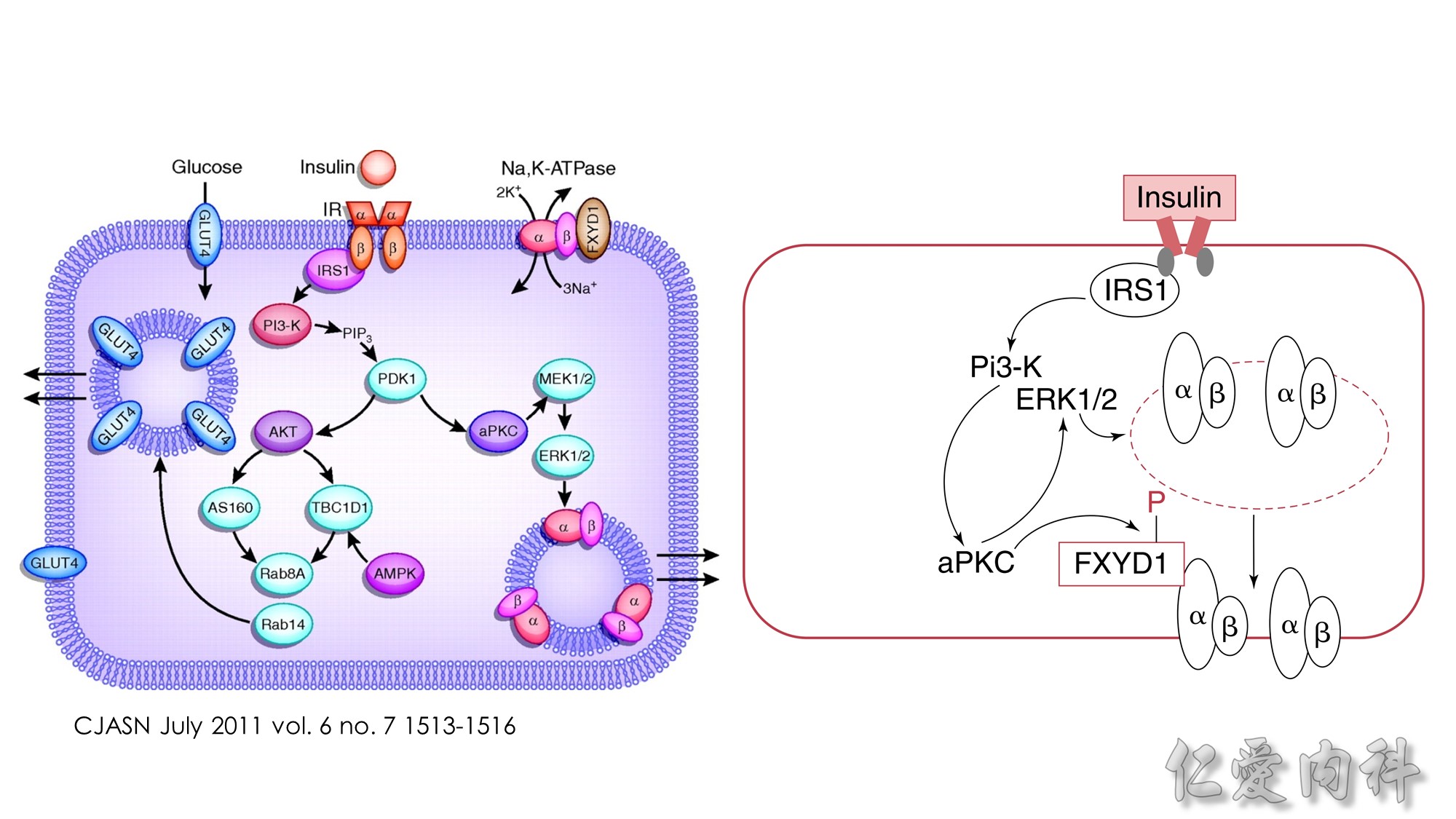 Insulin-stimulated glucose and potassium transport in skeletal muscle.
Insulin-stimulated glucose and potassium transport in skeletal muscle.
The figure summarizes the combined major signaling pathways and components involved in insulin-stimulated glucose and potassium cellular uptake by the facilitative glucose transporter GLUT4 and the Na,K-ATPase pump, respectively, in skeletal muscle. Negative regulatory pathways are not shown for clarity. Insulin binding to the insulin receptor, IR, a receptor tyrosine kinase consisting of a tetramer of extracellular α-subunits and intracellular β-subunits activates β-subunit kinase activity and transphosphorylation leading to phosphorylation of the insulin receptor substrate protein IRS-1. IRS proteins are characterized by pleckstrin-homology and phosphotyrosine-binding domains that enable the binding of IRS-1 to PI3-K, a dimer of catalytic and regulatory subunits. The IRS-1–PI3-K interaction generates phosphatidylinositol-3,4,5-triphosphate (PIP3), leading to activation of Akt/protein kinase B via the 3-phosphoinositide–dependent protein kinase 1 (PDK1). Akt/protein kinase B inhibits the Rab-GTPase–activating protein AS160, resulting in activation of Rab small GTPases required for translocation of GLUT4 from GLUT4 storage vesicles (GSVs) enriched in IRAP and the vesicle-associated SNARE protein VAMP-2. Docking and fusion of GSVs with the plasma membrane is regulated by insulin. GLUT4 and Na,K-ATPase do not co-localize to the same skeletal muscle intracellular membrane vesicles. Insulin increases Na,K-ATPase activity and plasma membrane expression through a PI3-K, atypical protein kinase C (aPKC), and ERK1/2 mitogen-activated protein kinase pathway, although the pathway is less well defined. ERK1/2 activation occurs through aPKC in a MEK1/2 kinase–dependent manner. ERK1/2 kinase phosphorylation of the Na,K-ATPase α-subunit promotes membrane expression and translocation of Na,K-ATPase from an intracellular membrane pool to the sarcolemma possibly in part by retarding clathrin-dependent endocytosis. The regulatory protein FXYD1, or phospholemman, belongs to a family of proteins characterized by an FXYD sequence that associates with Na,K-ATPase and modulates its affinities for Na+ or K+or its Imax. FXYD1 suppresses Na,K-ATPase activity by reducing its Na+affinity or altering its Imax. Phosphorylation of FXYD1 by protein kinase A or protein kinase C modifies its interaction with Na,K-ATPase.

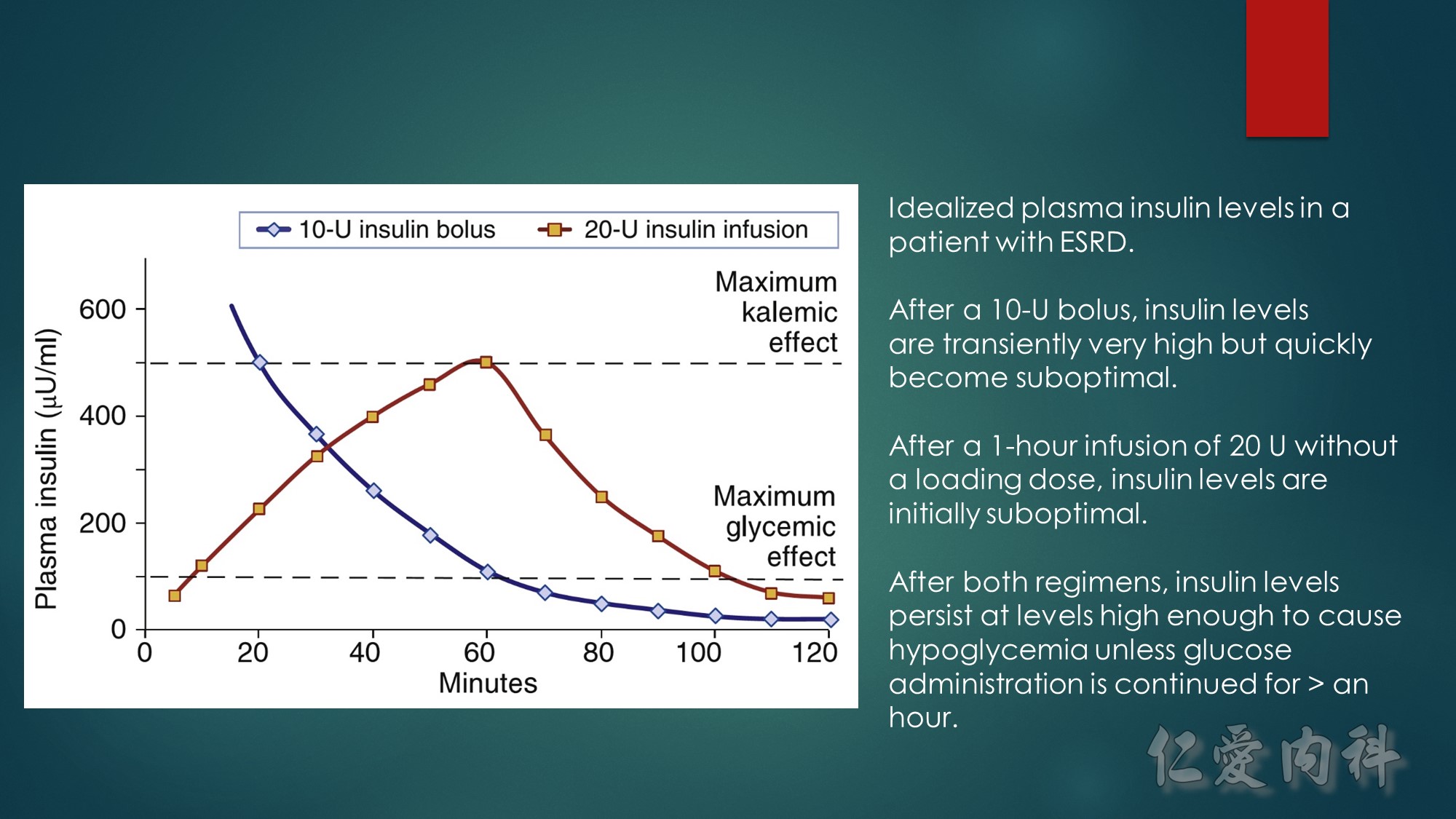

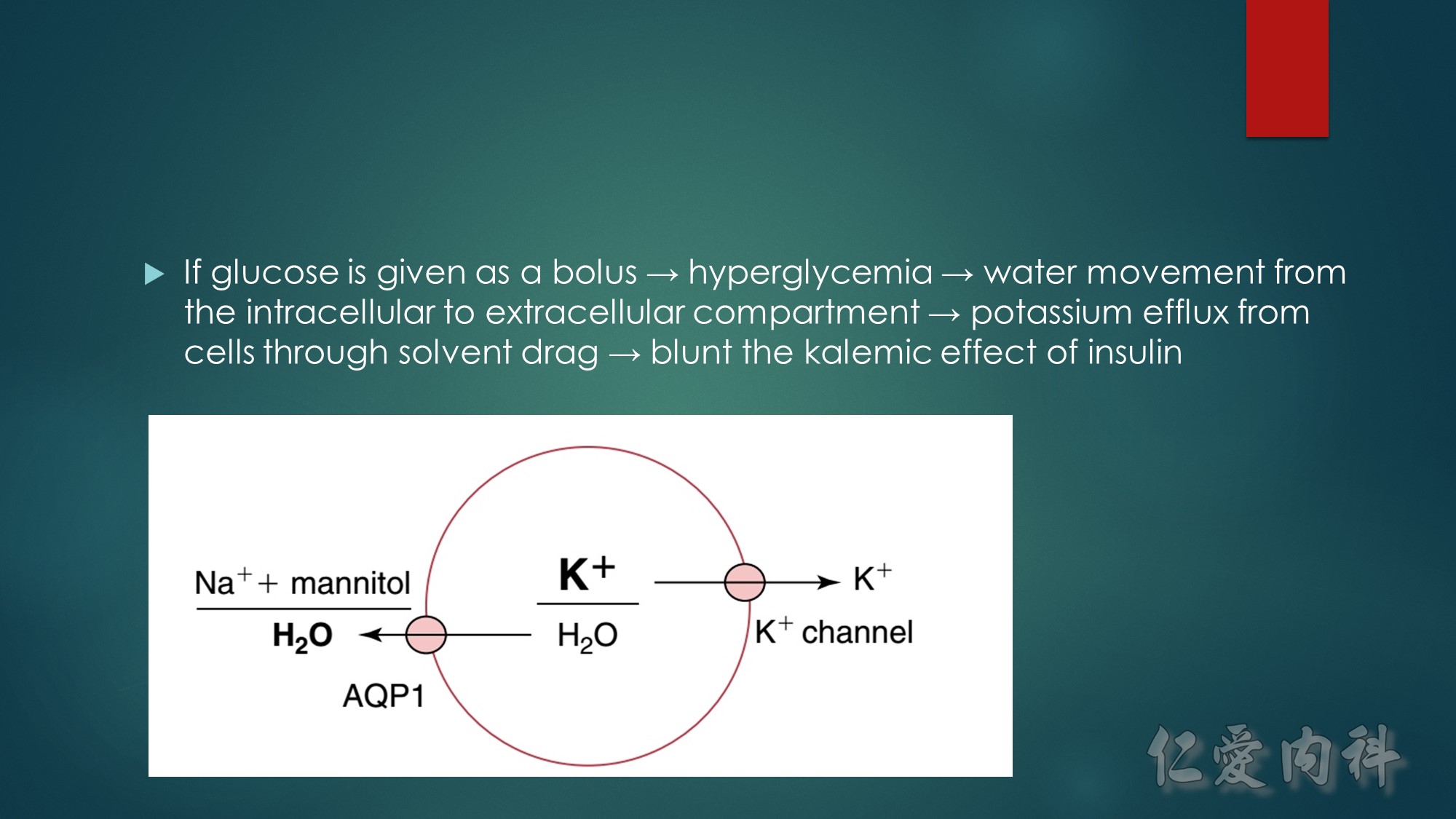


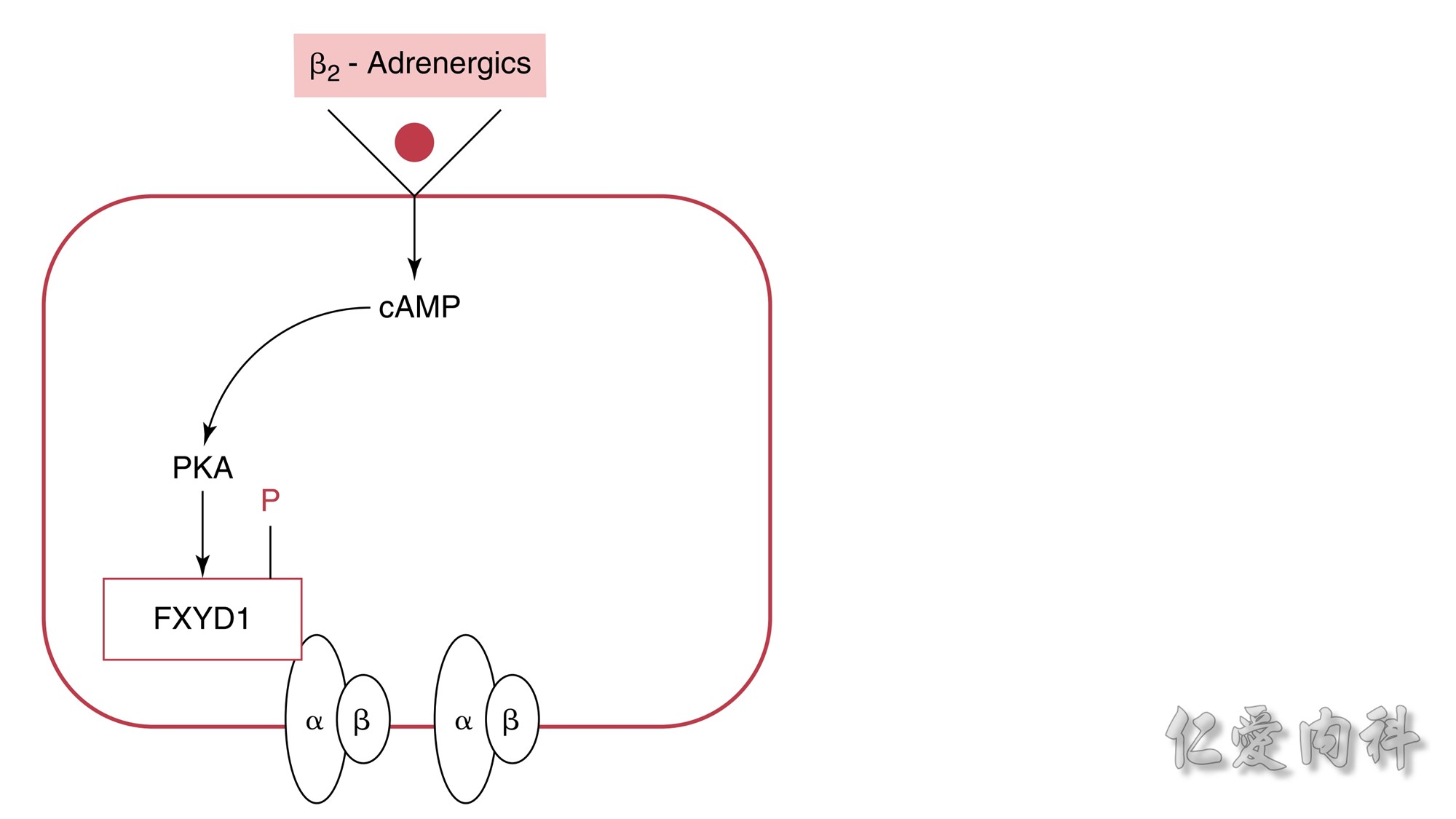 Figure 13-6 Mechanism of the Effect of β2-Adrenergic Agonists to Cause
Figure 13-6 Mechanism of the Effect of β2-Adrenergic Agonists to Cause
a Shift of K+ Ions into Cells.The rectangle represents a cell membrane. β2-
Adrenergic agonists activate adenylate cyclase, and this leads to stimulation of the conversion of adenosine triphosphate (ATP) to cyclic adenosine
monophosphate (cAMP). This second messenger, in turn, activates protein kinase-A (PKA), which induces phosphorylation of the FXYD1 (indicated by
the letter P in the figure). This disrupts the interaction of FXD1 with the α-subunit of the Na-K-ATPase, which results in an increase in the affinity
of Na-K-ATPase for Na+ ions and/or its Vmax. The increase in export of preexisting intracellular Na+ ions leads to a higher negative voltage in cells and
hence a shift of K+ ions into cells.















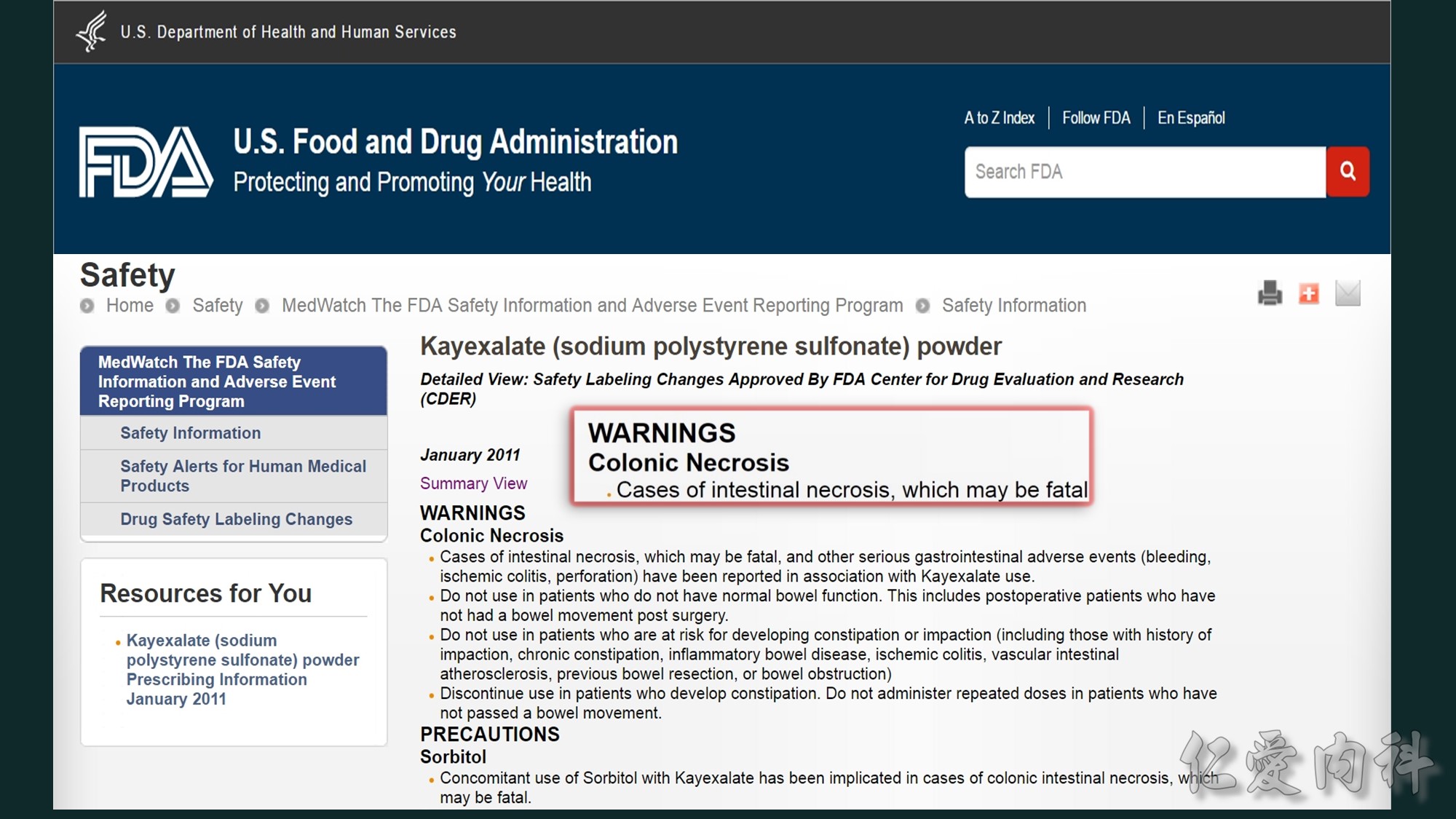
Sorbitol與polystyrene sulfonate併用會增加患者結腸壞死與上腸胃道損傷的風險。


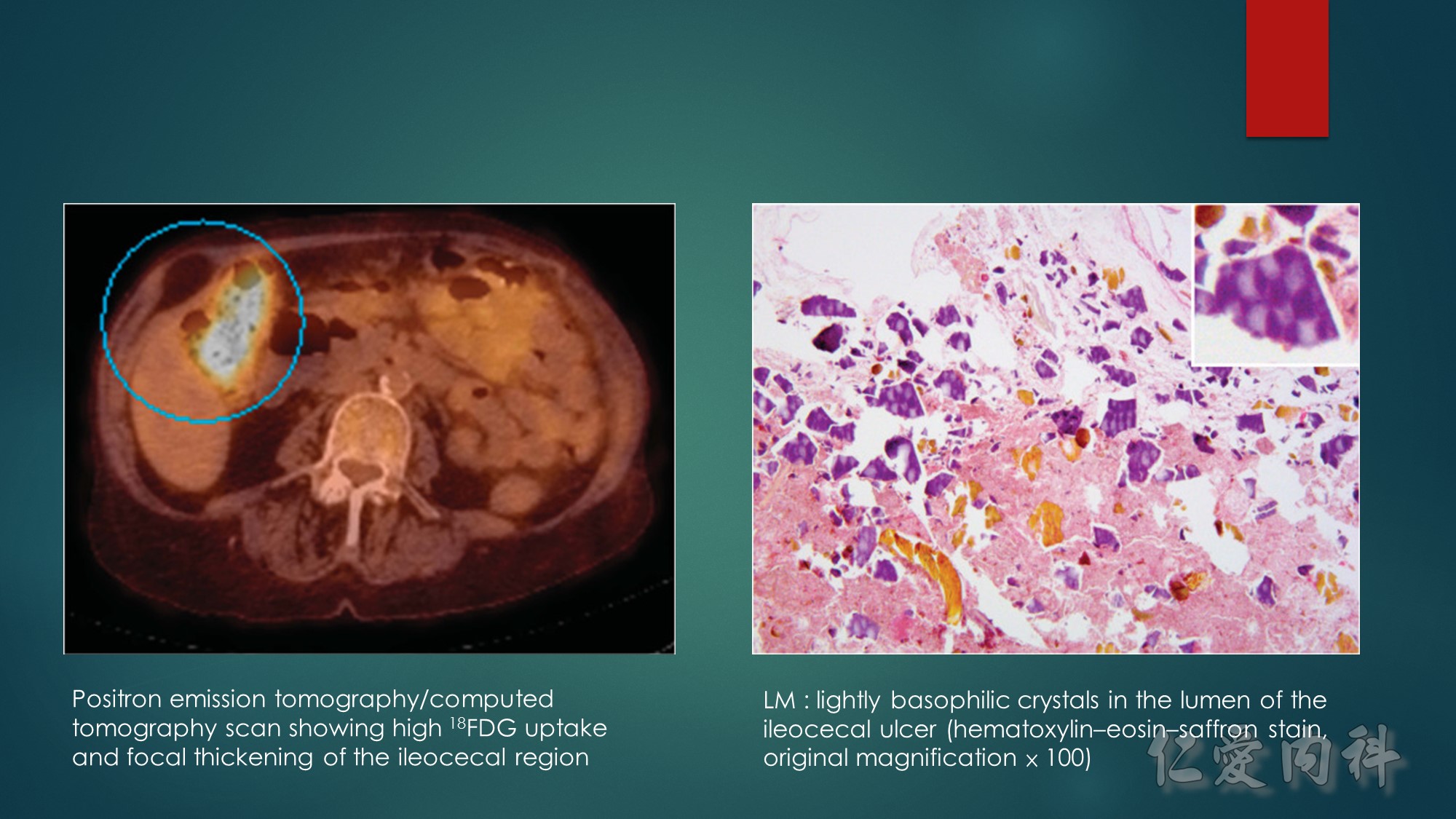
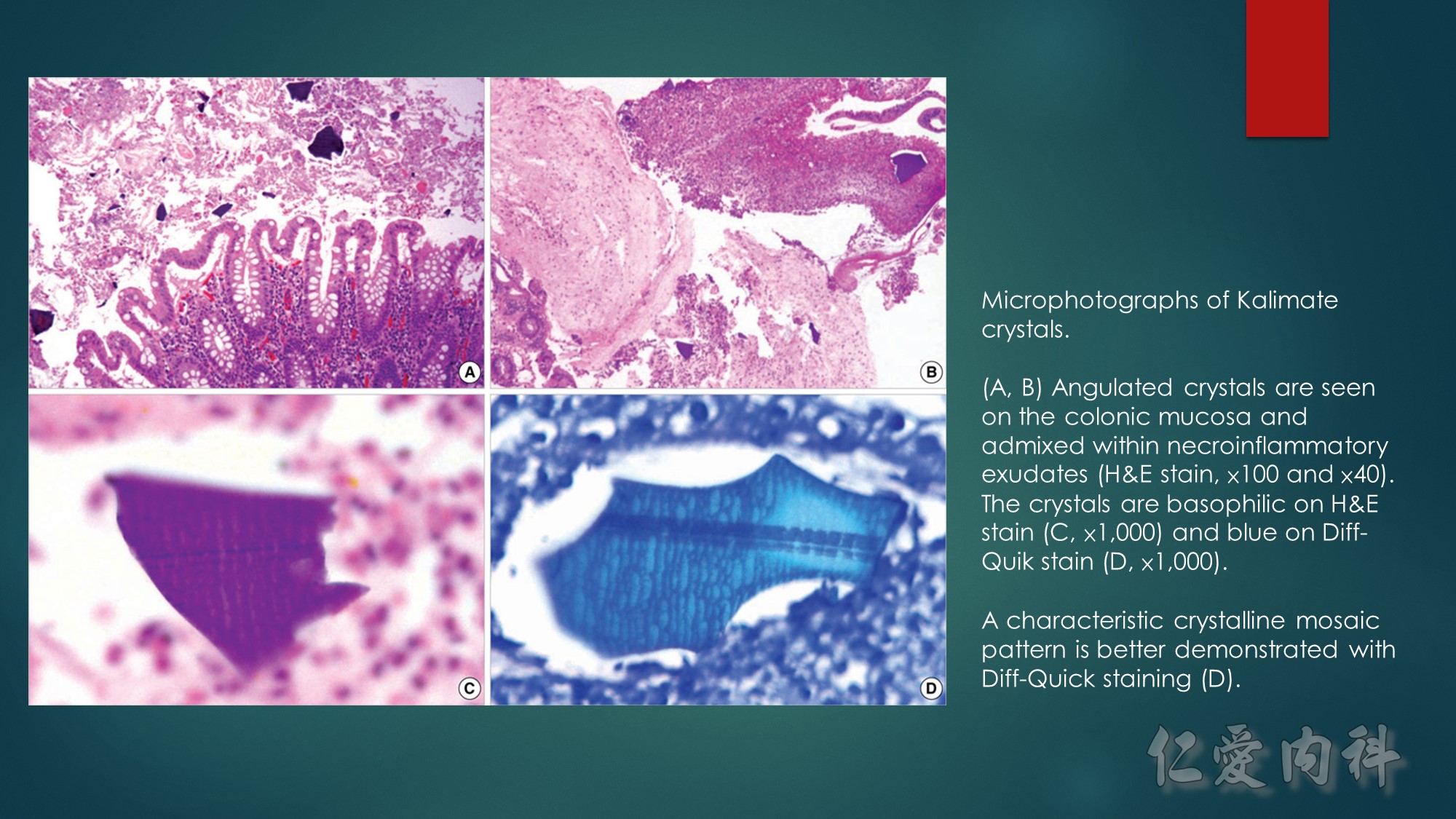
The abdominal computed tomography scan showed an infiltration of pericecal fat, and focal thickening and inflammation of the ileocecal region, with high uptake of 18FDG on 18FDG-PET/CT scan (Figure 1). On exploratory laparotomy, the ileocecal region was thickened and inflamed with enlarged lymph nodes. A right hemicolectomy and lymph node resection were undertaken. Gross examination showed a large ileal ulcer of 6 × 4.5cm and a calcified foreign body of 2 × 0.3cm. Lightly basophilic and purple polygonal crystals were observed on hematoxylin–eosin–saffron stain
SPS-related intestinal necrosis and other serious gastrointestinal events have been reported mainly in small case series.2., 3., 4. The incidence and relative risk are difficult to assess considering the need for large cohort studies.5 SPS-associated complications predominate on the ileum and colon, but may affect the upper gastrointestinal tract and include bleeding, ischemic colitis, focal to deep ulceration, necrosis and perforation, and fecal impaction with rectal stenosis.2., 3., 4. Reported risk factors include prematurity, history of intestinal disease, surgery or transplantation, hypovolemia, and renal failure.1,2 It is noteworthy that sorbitol, a cathartic often added to prevent obstipation, has been incriminated to trigger SPS-related serious gastrointestinal events.
This case of particularly severe foreign-body reaction illustrates an unexpected cause of severe weight loss, fever, and abdominal pain in the renal transplant recipient, and a potential new aspect of SPS-associated serious gastrointestinal complications.




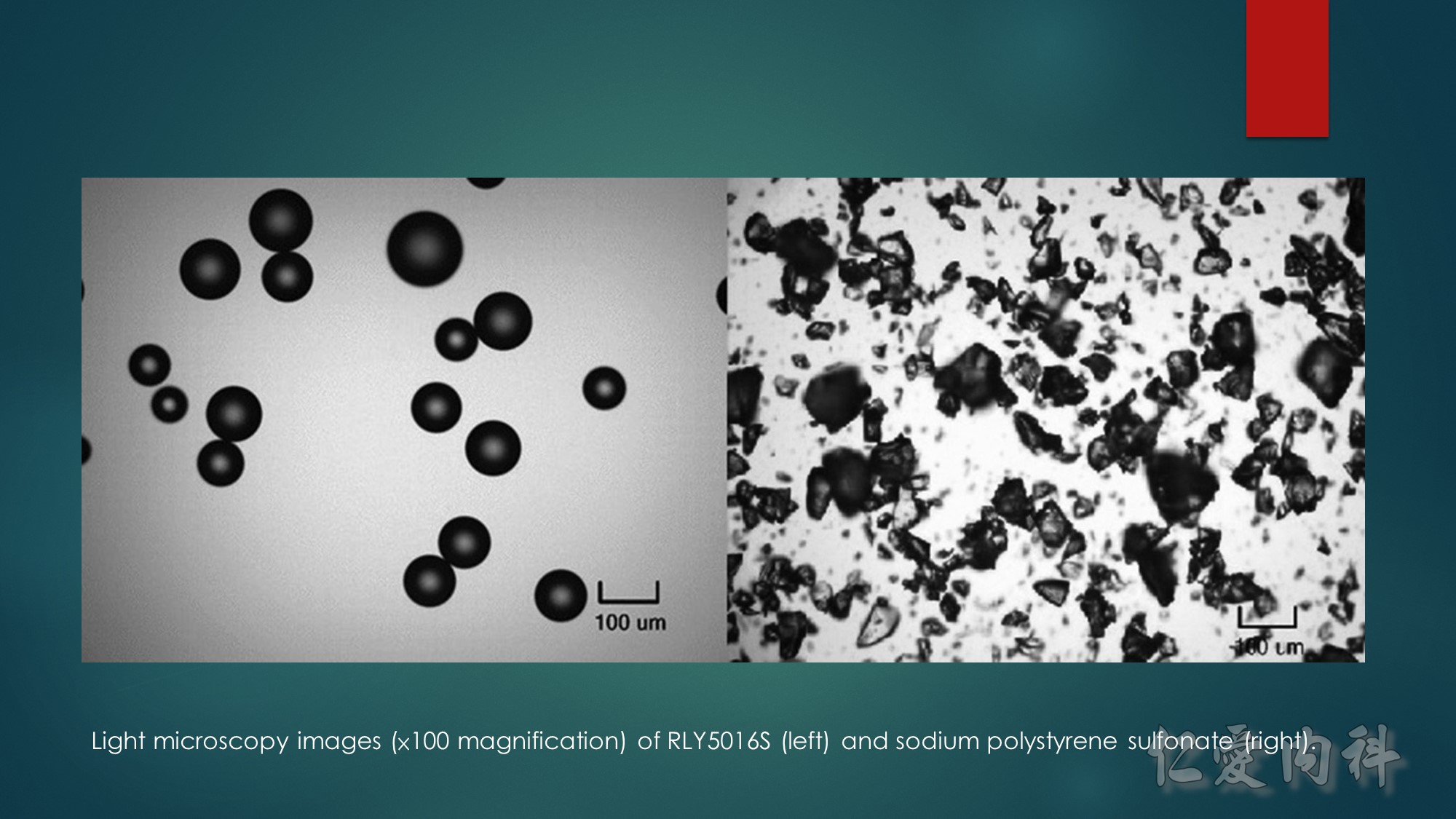
Light microscopy images (×100 magnification) of RLY5016S (left) and sodium polystyrene sulfonate (right).
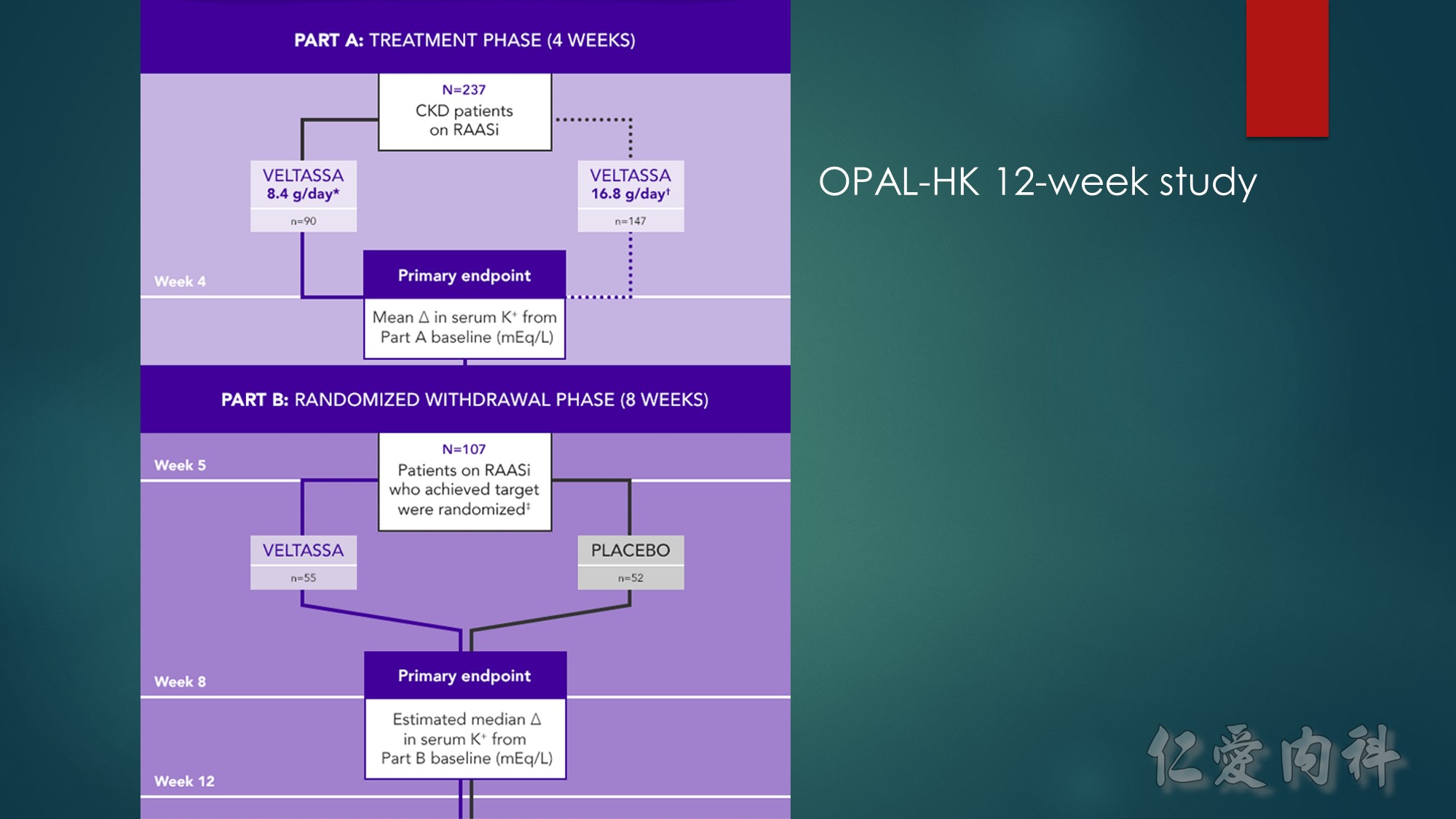
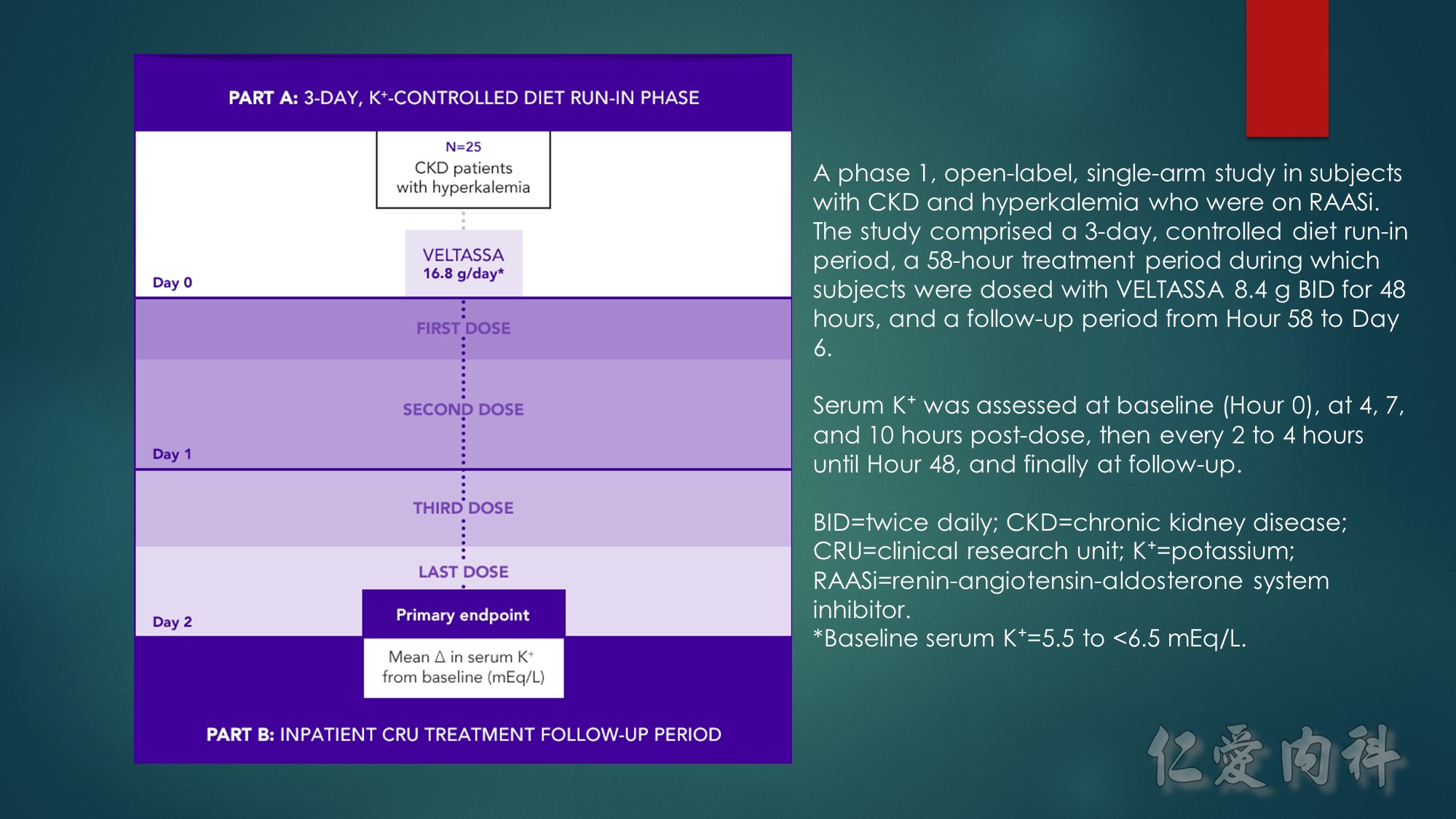
A phase 3, 2-part, 12-week, multicenter study consisting of a 4-week, single-group, single-blind initial treatment phase and an 8-week, placebo-controlled, single-blind, randomized withdrawal phase in adult patients with CKD stage 3 or 4. Patients had serum K⁺ of 5.1 to <6.5 mEq/L and were receiving a stable dose of RAASi at screening. During the initial treatment phase, patients with mild hyperkalemia received an initial dose of VELTASSA 8.4 g/day (as a divided dose), and those with moderate to severe hyperkalemia received VELTASSA 16.8 g/day (as a divided dose).
CKD=chronic kidney disease; K⁺=potassium; RAASi=renin-angiotensin-aldosterone system inhibitor.
*Baseline serum K⁺=5.1 to <5.5 mEq/L.
†Baseline serum K⁺=5.5 to <6.5 mEq/L.
‡Target serum K⁺=3.8 to <5.1 mEq/L.
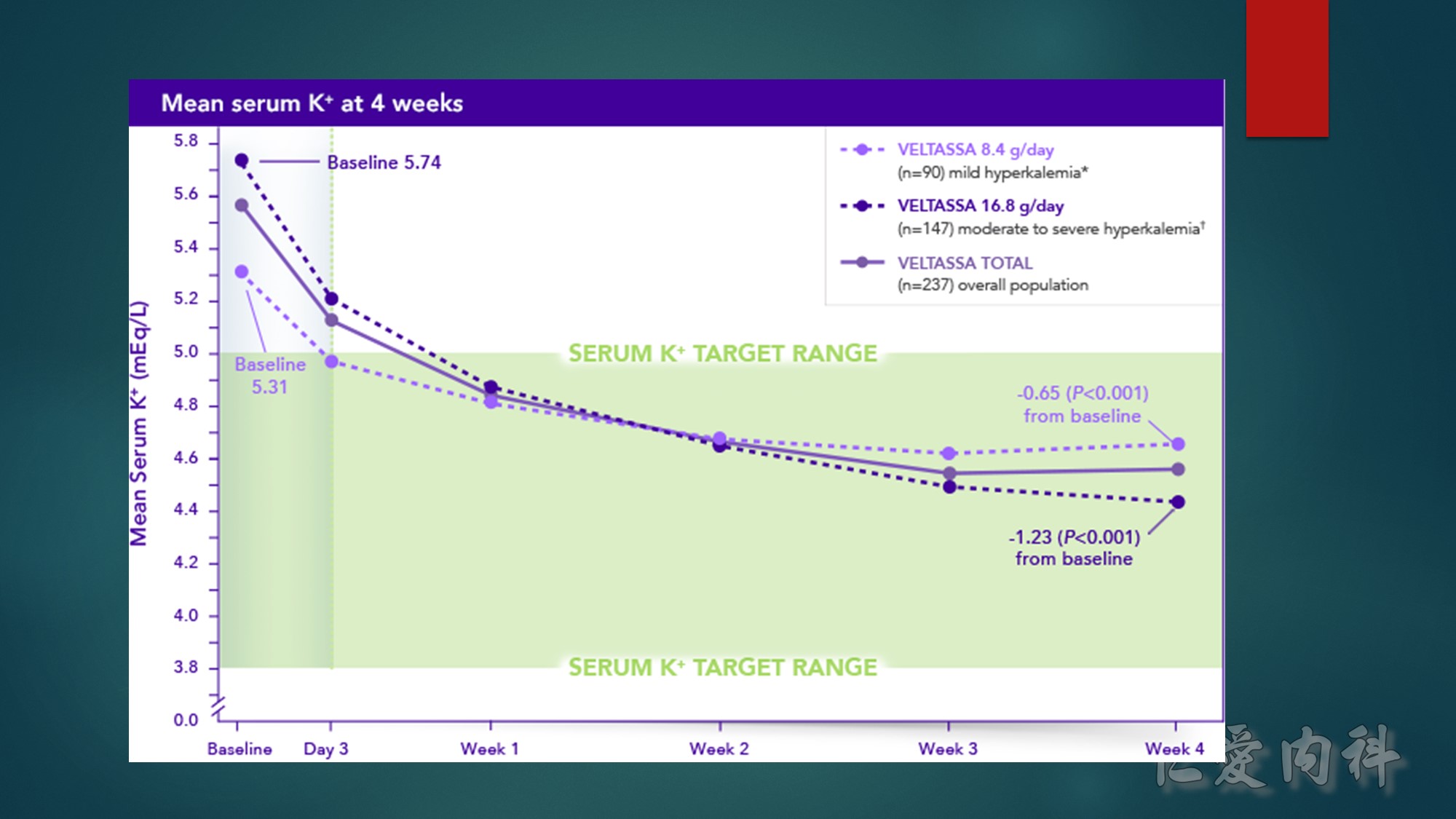
Incidence of hypokalemia was 3% and often transient, resolving with dose adjustment.
*Baseline serum K⁺=5.1 to <5.5 mEq/L.
†Baseline serum K⁺=5.5 to <6.5 mEq/L.
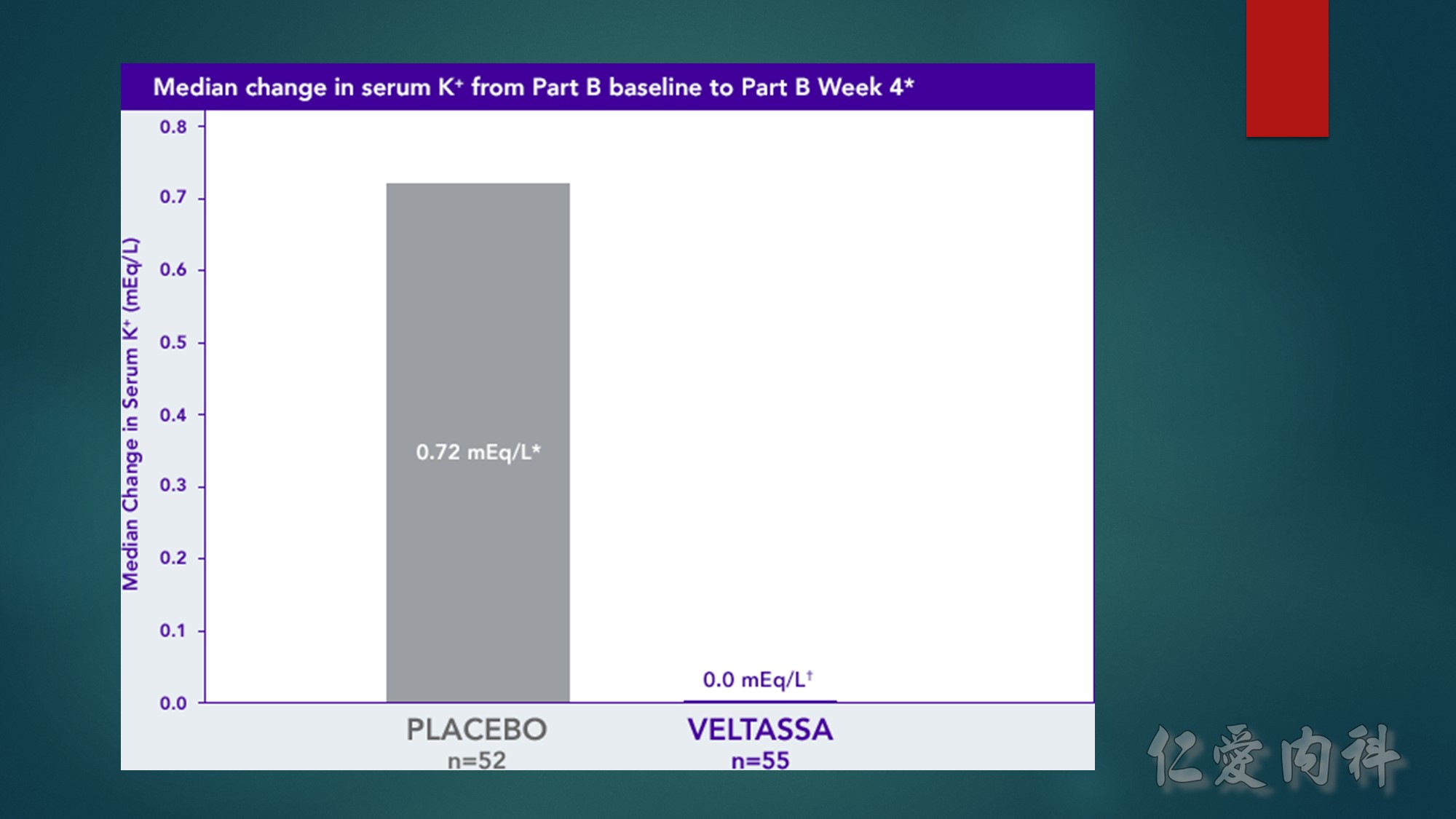
The median serum K⁺ of the VELTASSA group remained stable from Week 4 to Week 8, while the placebo group had an increase in median serum K⁺ of 0.72 mEq/L.†
A total of 60% of the patients in the placebo group vs 15% in the VELTASSA group had at least one recurrence of moderate to severe hyperkalemia through Week 8 of the withdrawal phase.†‡
K⁺=potassium.
*The Part B primary endpoint was the change in serum K⁺ from Part B baseline to the earliest visit at which the patient’s serum K⁺ was first outside of the range of 3.8 to <5.5 mEq/L, or to Part B Week 4 if the patient’s serum K⁺ remained in the range.
†P<0.001.
‡Serum K⁺=≥5.5 mEq/L.
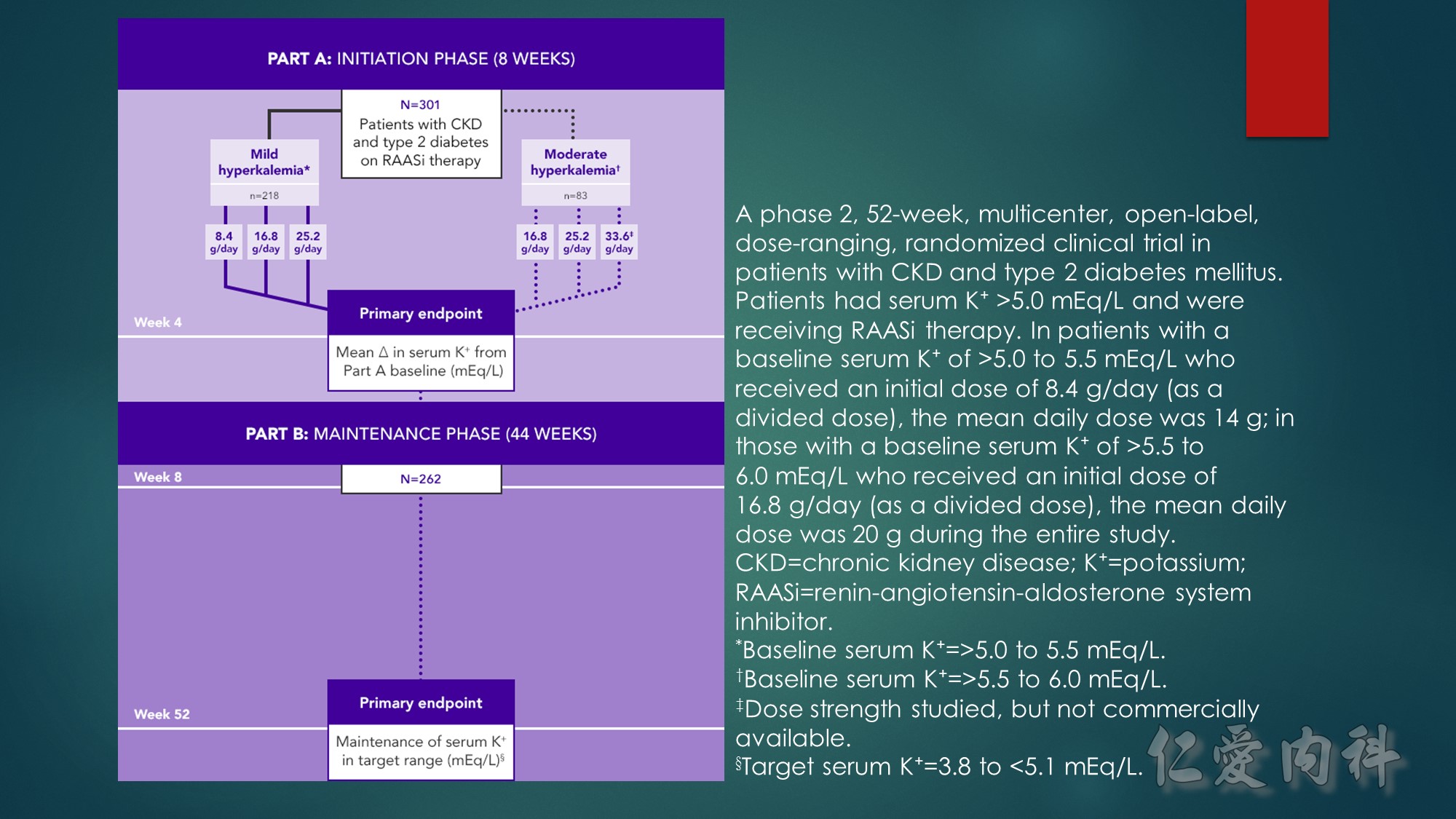
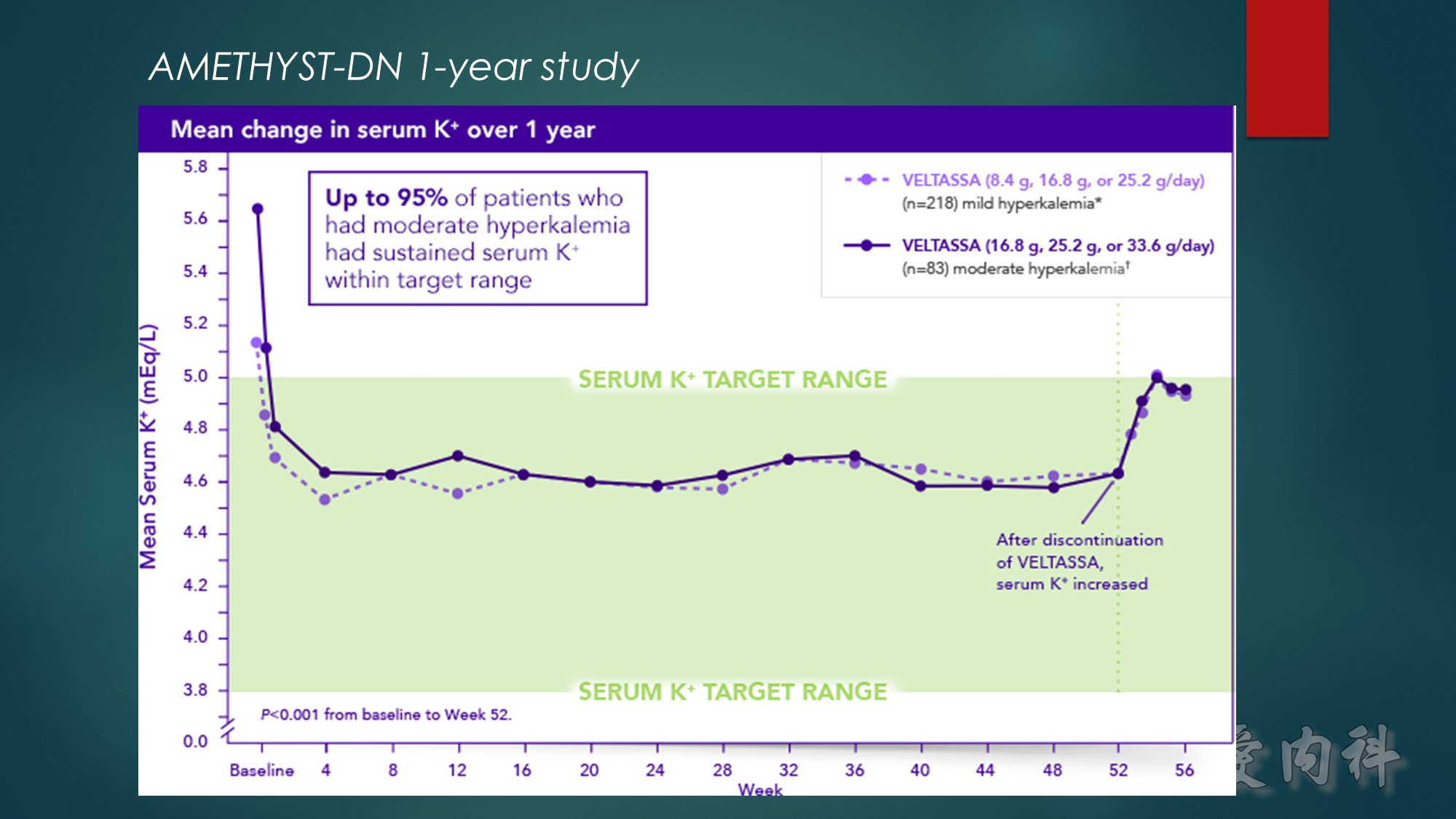
Hypokalemia occurred in 5.6% of patients over 52 weeks, with no patients developing serum K⁺ <3.0 mEq/L
Approximately 69% of all patients studied completed treatment at 52 weeks
*Baseline serum K⁺=>5.0 to 5.5 mEq/L.
†Baseline serum K⁺=>5.5 to 6.0 mEq/L.
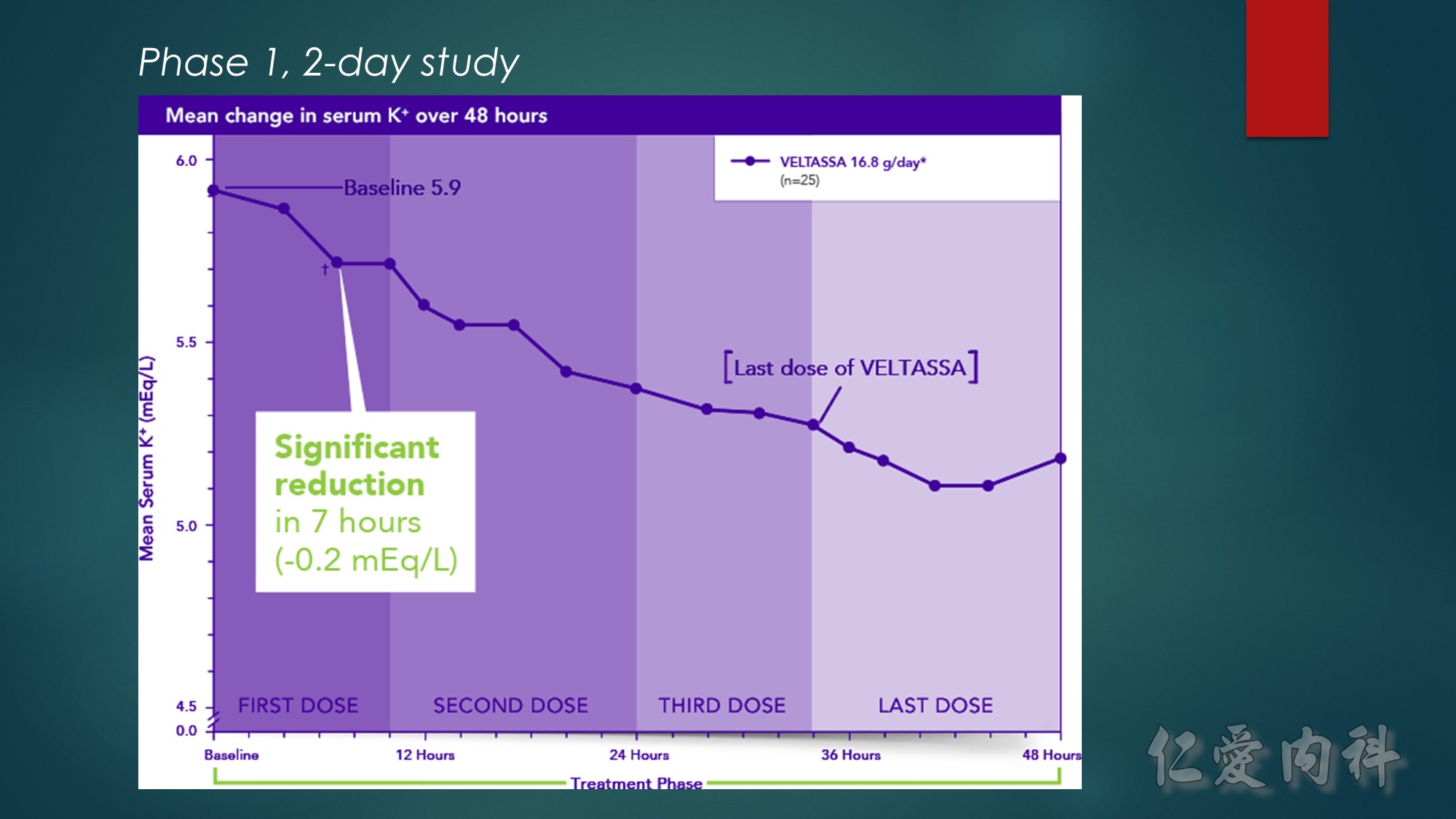
*Baseline serum K⁺=5.5 to <6.5 mEq/L.
†P≤0.004.




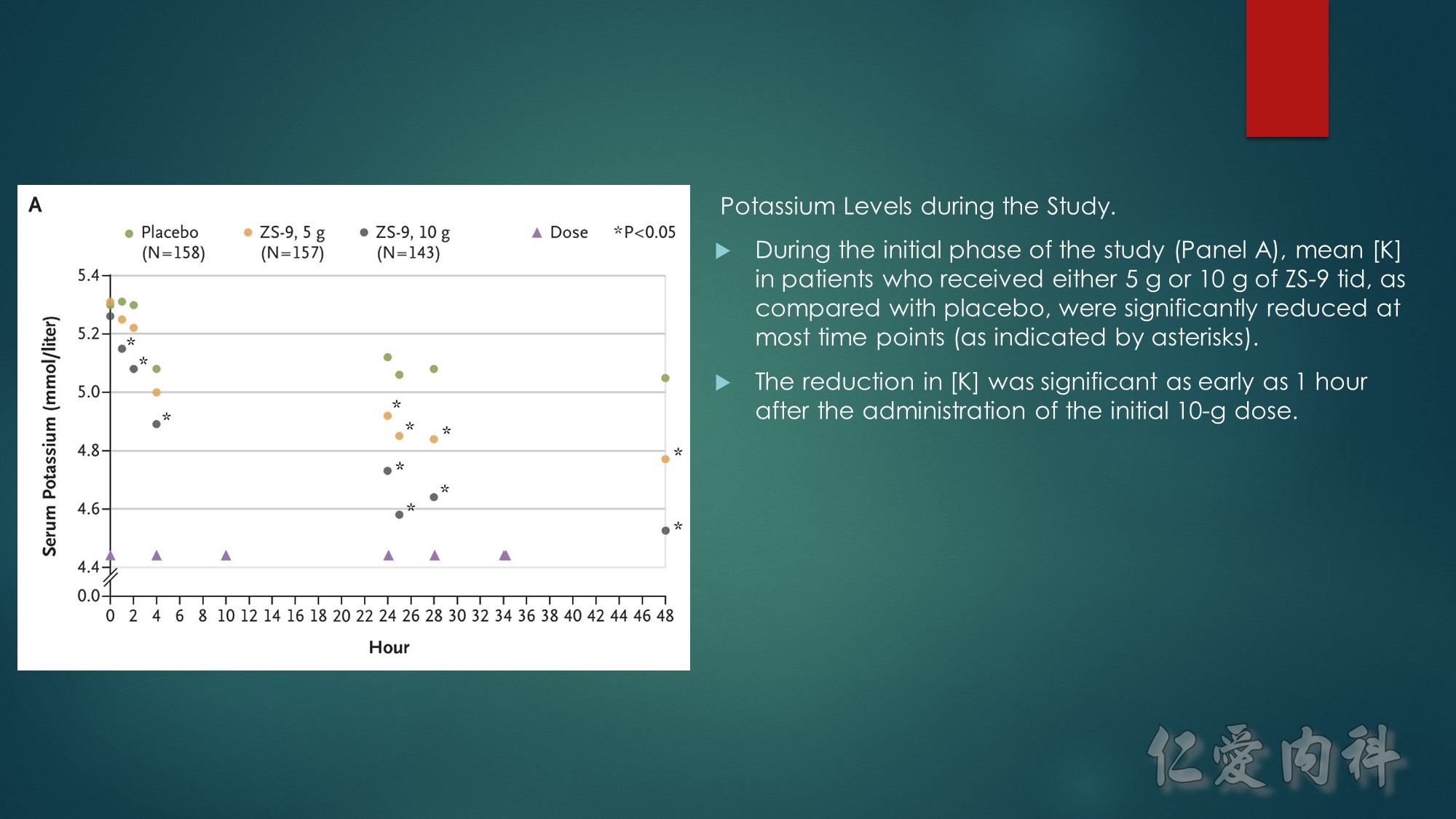
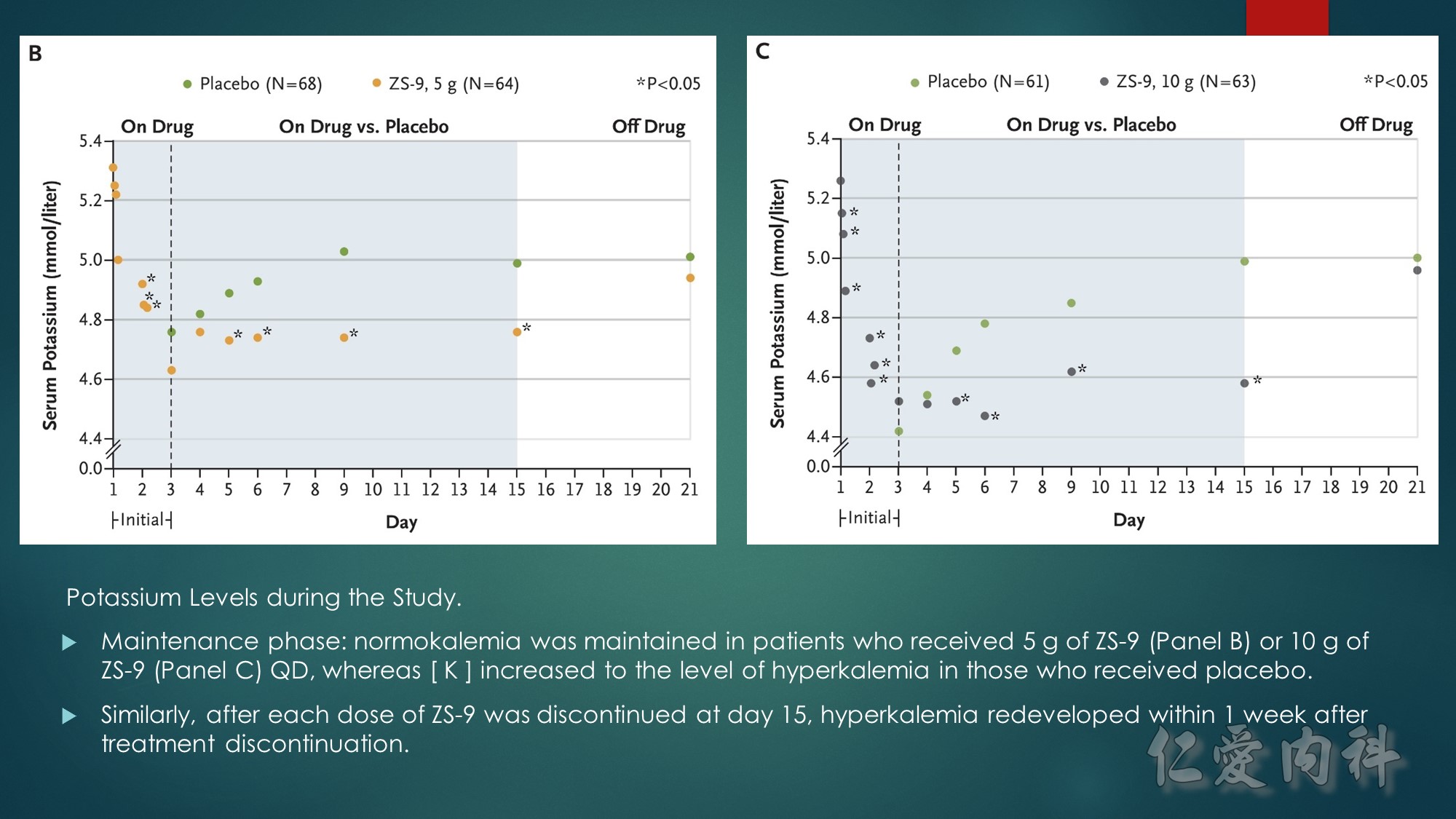

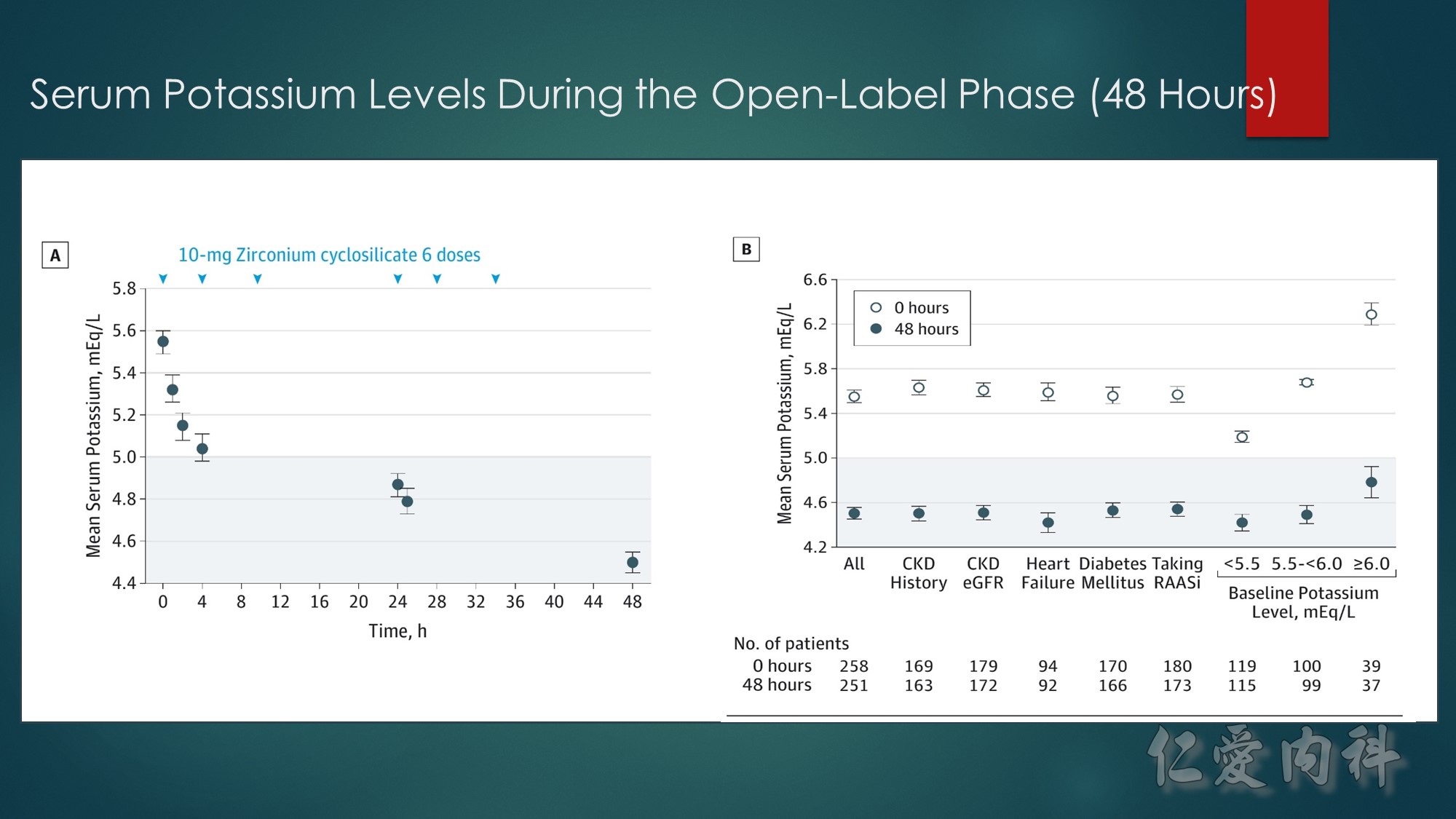
A, Mean serum potassium levels over time in patients treated during the open-label phase with zirconium cyclosilicate, 10 g, 3 times daily for 48 hours.
B, Mean serum potassium levels at 0 and 48 hours across prespecified subgroups of chronic kidney disease (CKD) (by patient history and by estimated glomerular filtration rate [eGFR]

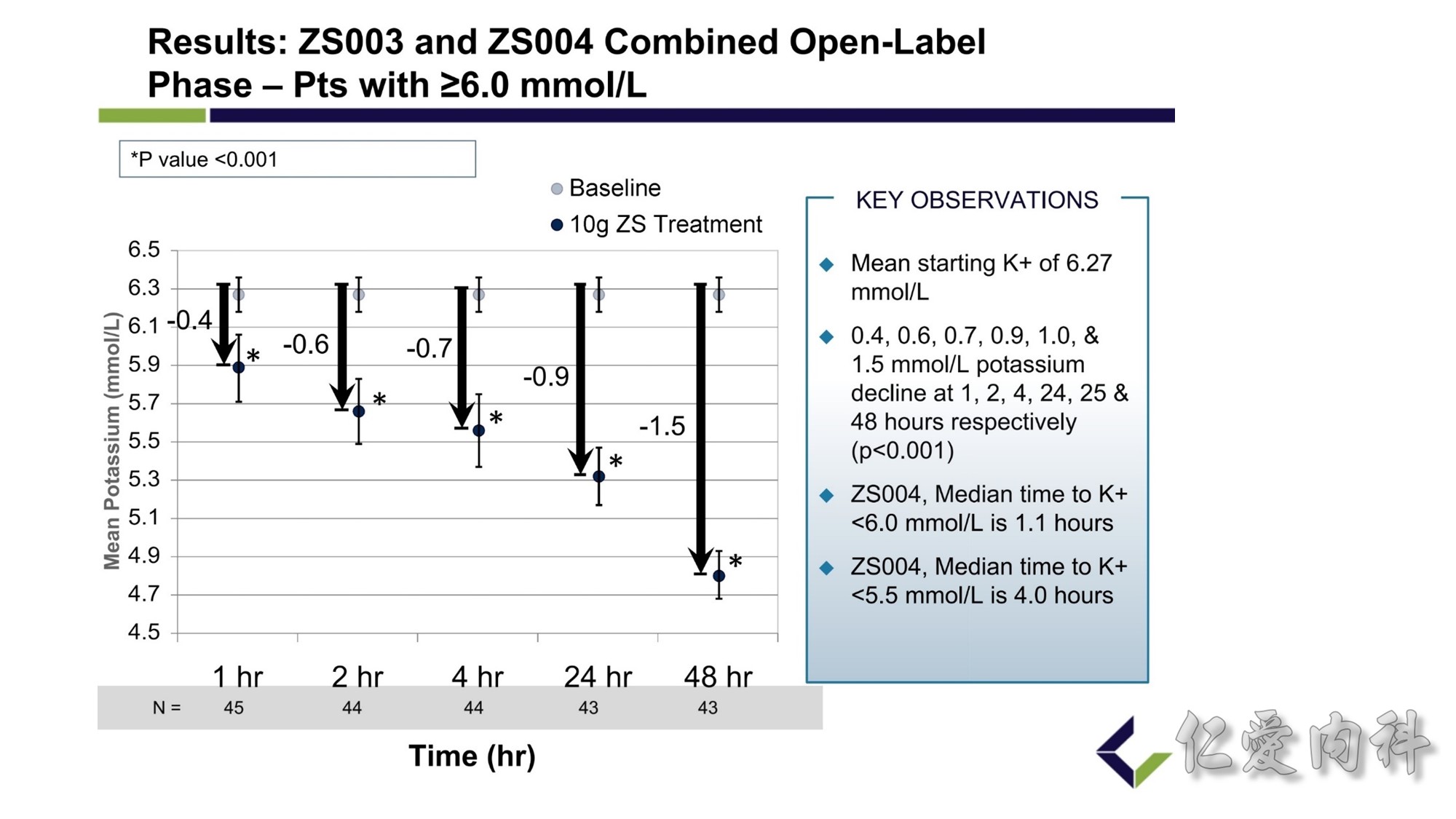
In a subgroup of 45 patients with serum potassium concentrations of at least 6 mmol/l (6.1 to 7.2 mmol/l), who participated in 2 controlled trials, administration of 10 g of ZS-9 significantly reduced the serum potassium concentration below baseline by 0.4 mmol/l at 1 hour, by 0.6 mmol/l at 2 hours, and by 0.7 mmol/l at 4 hours (P < 0.001).